Oncogenesis ( IF 6.2 ) Pub Date : 2023-02-17 , DOI: 10.1038/s41389-023-00454-6 Fabio Vanoli 1 , Laurie Herviou 1 , Yusuke Tsuda 1 , Patricia Sung 1 , Ziyu Xie 1 , Eve Fishinevich 1 , Soe S Min 1 , William Mallen 1 , Henry de Traux de Wardin 1 , Yanming Zhang 1 , Maria Jasin 2 , Cristina R Antonescu 1
|
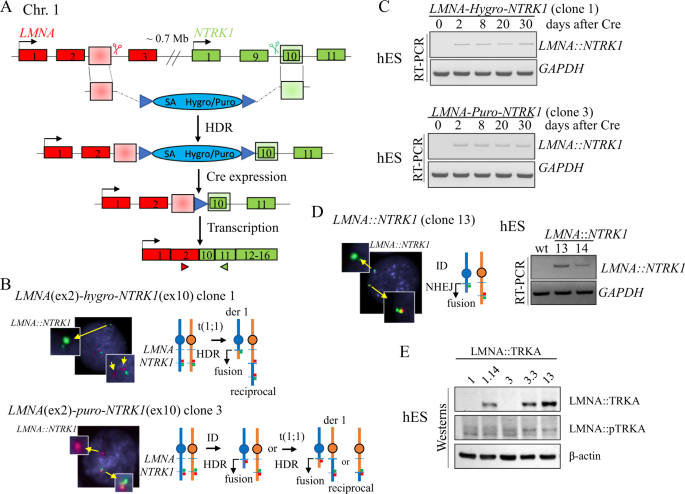
The discovery of neurotrophic tyrosine receptor kinase (NTRK) gene fusions as pan-tumor oncogenic drivers has led to new personalized therapies in oncology. Recent studies investigating NTRK fusions among mesenchymal neoplasms have identified several emerging soft tissue tumor entities displaying various phenotypes and clinical behaviors. Among them, tumors resembling lipofibromatosis or malignant peripheral nerve sheath tumors often harbor intra-chromosomal NTRK1 rearrangements, while most infantile fibrosarcomas are characterized by canonical ETV6::NTRK3 fusions. However, appropriate cellular models to investigate mechanisms of how kinase oncogenic activation through gene fusions drives such a wide spectrum of morphology and malignancy are lacking. Progress in genome editing has facilitated the efficient generation of chromosomal translocations in isogenic cell lines. In this study we employ various strategies to model NTRK fusions, including LMNA::NTRK1 (interstitial deletion) and ETV6::NTRK3 (reciprocal translocation) in human embryonic stem (hES) cells and mesenchymal progenitors (hES-MP). Here, we undertake various methods to model non-reciprocal, intrachromosomal deletions/translocations by induction of DNA double strand breaks (DSBs) exploiting either the repair mechanisms of homology directed repair (HDR) or non-homologous end joining (NHEJ). Expression of LMNA::NTRK1 or ETV6::NTRK3 fusions in either hES cells or hES-MP did not affect cell proliferation. However, the level of mRNA expression of the fusion transcripts was significantly upregulated in hES-MP, and phosphorylation of the LMNA::NTRK1 fusion oncoprotein was noted only in hES-MP but not in hES cells. Similarly, an NTRK1-driven transcriptional profile related to neuronal and neuroectodermal lineage was upregulated mainly in hES-MP, supporting the importance of appropriate cellular context in modeling cancer relevant aberrations. As proof of concept of the validity of our in vitro models, phosphorylation was depleted by two TRK inhibitors, Entrectinib and Larotrectinib, currently used as targeted therapy for tumors with NTRK fusions.
中文翻译:
生成 NTRK 融合间充质瘤的体外模型作为研究激酶致癌激活和靶向治疗反应的工具
作为泛肿瘤致癌驱动因素的神经营养性酪氨酸受体激酶 (NTRK) 基因融合的发现导致了肿瘤学中新的个性化疗法。最近调查间充质肿瘤中NTRK融合的研究已经确定了几种新出现的软组织肿瘤实体,这些实体显示出各种表型和临床行为。其中,类似于脂肪纤维瘤病或恶性周围神经鞘瘤的肿瘤通常具有染色体内NTRK1重排,而大多数婴儿纤维肉瘤的特征在于典型的ETV6::NTRK3融合。然而,缺乏适当的细胞模型来研究激酶致癌激活如何通过基因融合驱动如此广泛的形态学和恶性肿瘤的机制。基因组编辑的进展促进了同基因细胞系中染色体易位的有效产生。在这项研究中,我们采用各种策略来模拟NTRK融合,包括LMNA::NTRK1(间质删除)和ETV6::NTRK3(相互易位)在人胚胎干细胞 (hES) 和间充质祖细胞 (hES-MP) 中。在这里,我们采用各种方法通过利用同源定向修复 (HDR) 或非同源末端连接 (NHEJ) 的修复机制诱导 DNA 双链断裂 (DSB) 来模拟非互惠的染色体内缺失/易位。LMNA::NTRK1或ETV6::NTRK3融合体在 hES 细胞或 hES-MP 中的表达不影响细胞增殖。然而,融合转录物的 mRNA 表达水平在 hES-MP 中显着上调,并且LMNA::NTRK1融合癌蛋白的磷酸化仅在 hES-MP 中而非 hES 细胞中注意到。同样,一个NTRK1与神经元和神经外胚层谱系相关的驱动转录谱主要在 hES-MP 中上调,支持适当的细胞环境在模拟癌症相关畸变中的重要性。作为我们体外模型有效性的概念证明,磷酸化被两种 TRK 抑制剂 Entrectinib 和 Larotrectinib 耗尽,目前用作NTRK融合肿瘤的靶向治疗。


























 京公网安备 11010802027423号
京公网安备 11010802027423号