Neurogenetics ( IF 2.2 ) Pub Date : 2023-05-05 , DOI: 10.1007/s10048-023-00718-8 Viviana Tritto 1 , Federico Grilli 2 , Donatella Milani 2 , Paola Riva 1
|
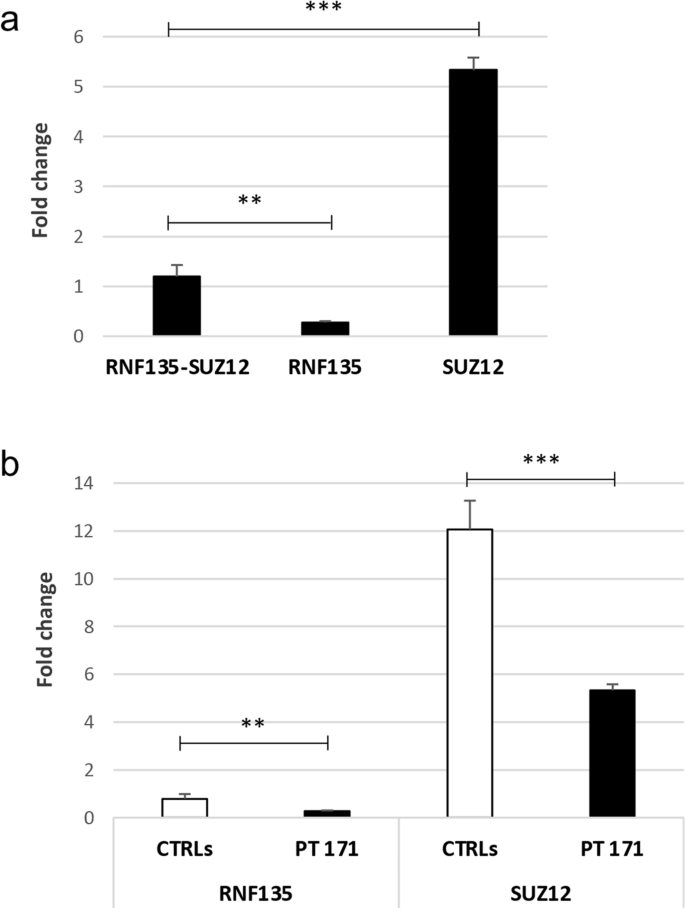
Neurofibromatosis type I (NF1) microdeletion syndrome, accounting for 5–11% of NF1 patients, is caused by the heterozygous deletion of NF1 and a variable number of flanking genes in the 17q11.2 region. This syndrome is characterized by more severe symptoms than those shown by patients with intragenic NF1 mutation and by variable expressivity, which is not fully explained by the haploinsufficiency of the genes included in the deletions. We here reevaluate an 8-year-old NF1 patient, who carries an atypical deletion generating the RNF135-SUZ12 chimeric gene, previously described when he was 3 years old. As the patient has developed multiple cutaneous/subcutaneous neurofibromas over the past 5 years, we hypothesized a role of RNF135-SUZ12 chimeric gene in the onset of the patient’s tumor phenotype. Interestingly, SUZ12 is generally lost or disrupted in NF1 microdeletion syndrome and frequently associated to cancer as RNF135. Expression analysis confirmed the presence of the chimeric gene transcript and revealed hypo-expression of five out of the seven analyzed target genes of the polycomb repressive complex 2 (PRC2), to which SUZ12 belongs, in the patient’s peripheral blood, indicating a higher transcriptional repression activity mediated by PRC2. Furthermore, decreased expression of tumor suppressor gene TP53, which is targeted by RNF135, was detected. These results suggest that RNF135-SUZ12 chimera may acquire a gain of function, compared with SUZ12 wild type in the PRC2 complex, and a loss of function relative to RNF135 wild type. Both events may have a role in the early onset of the patient’s neurofibromas.
中文翻译:
NF1 患者中多梳抑制复合物 2 靶基因的表达失调,微缺失产生 RNF135-SUZ12 嵌合基因
I 型神经纤维瘤病 (NF1) 微缺失综合征占 NF1 患者的 5-11%,是由NF1杂合缺失和 17q11.2 区域中不同数量的侧翼基因引起的。该综合征的特点是比基因内NF1突变患者表现出的症状更严重,并且表达性可变,这不能完全用缺失中包含的基因的单倍体不足来解释。我们在此重新评估一名 8 岁的 NF1 患者,该患者携带产生RNF135-SUZ12嵌合基因的非典型缺失,此前在他 3 岁时曾描述过该基因。由于患者在过去 5 年中出现了多发性皮肤/皮下神经纤维瘤,我们假设RNF135-SUZ12嵌合基因在患者肿瘤表型的发生中发挥作用。有趣的是,SUZ12通常在 NF1 微缺失综合征中丢失或破坏,并且经常与癌症相关(如RNF135)。表达分析证实了嵌合基因转录本的存在,并揭示了患者外周血中 SUZ12 所属的多梳抑制复合物 2 (PRC2) 的 7 个分析靶基因中有 5 个低表达,表明转录抑制程度较高PRC2 介导的活性。此外,还检测到 RNF135 靶向的肿瘤抑制基因TP53的表达降低。这些结果表明,与PRC2复合物中的SUZ12野生型相比,RNF135-SUZ12嵌合体可能获得功能获得,并且相对于RNF135野生型获得功能丧失。这两种事件可能在患者神经纤维瘤的早期发病中发挥作用。


























 京公网安备 11010802027423号
京公网安备 11010802027423号