当前位置:
X-MOL 学术
›
Genes Brain Behav.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Spontaneous allelic variant in deafness–blindness gene Ush1g resulting in an expanded phenotype
Genes, Brain and Behavior ( IF 2.5 ) Pub Date : 2023-06-16 , DOI: 10.1111/gbb.12849 Vladimir Vartanian 1 , Jocelyn F Krey 2 , Paroma Chatterjee 2 , Allison Curtis 3 , Makayla Six 3 , Sean P M Rice 4 , Sherri M Jones 5 , Harini Sampath 6 , Charles N Allen 7 , Renee C Ryals 3 , R Stephen Lloyd 1, 8 , Peter G Barr-Gillespie 2
Genes, Brain and Behavior ( IF 2.5 ) Pub Date : 2023-06-16 , DOI: 10.1111/gbb.12849 Vladimir Vartanian 1 , Jocelyn F Krey 2 , Paroma Chatterjee 2 , Allison Curtis 3 , Makayla Six 3 , Sean P M Rice 4 , Sherri M Jones 5 , Harini Sampath 6 , Charles N Allen 7 , Renee C Ryals 3 , R Stephen Lloyd 1, 8 , Peter G Barr-Gillespie 2
Affiliation
|
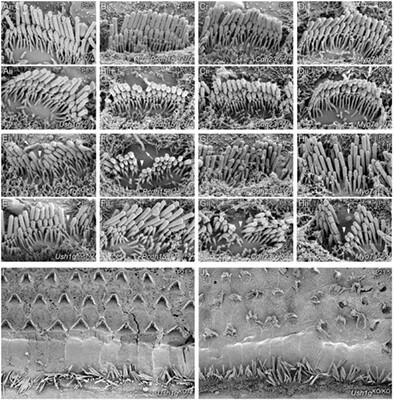
Relationships between novel phenotypic behaviors and specific genetic alterations are often discovered using target-specific, directed mutagenesis or phenotypic selection following chemical mutagenesis. An alternative approach is to exploit deficiencies in DNA repair pathways that maintain genetic integrity in response to spontaneously induced damage. Mice deficient in the DNA glycosylase NEIL1 show elevated spontaneous mutations, which arise from translesion DNA synthesis past oxidatively induced base damage. Several litters of Neil1 knockout mice included animals that were distinguished by their backwards-walking behavior in open-field environments, while maintaining frantic forward movements in their home cage environment. Other phenotypic manifestations included swim test failures, head tilting and circling. Mapping of the mutation that conferred these behaviors showed the introduction of a stop codon at amino acid 4 of the Ush1g gene. Ush1gbw/bw null mice displayed auditory and vestibular defects that are commonly seen with mutations affecting inner-ear hair-cell function, including a complete lack of auditory brainstem responses and vestibular-evoked potentials. As in other Usher syndrome type I mutant mouse lines, hair cell phenotypes included disorganized and split hair bundles, as well as altered distribution of proteins for stereocilia that localize to the tips of row 1 or row 2. Disruption to the bundle and kinocilium displacement suggested that USH1G is essential for forming the hair cell's kinocilial links. Consistent with other Usher type 1 models, Ush1gbw/bw mice had no substantial retinal degeneration compared with Ush1gbw/+ controls. In contrast to previously described Ush1g alleles, this new allele provides the first knockout model for this gene.
中文翻译:

耳聋失明基因 Ush1g 的自发等位基因变异导致表型扩大
新的表型行为和特定遗传改变之间的关系通常通过目标特异性、定向诱变或化学诱变后的表型选择来发现。另一种方法是利用 DNA 修复途径的缺陷来维持遗传完整性,以应对自发引起的损伤。DNA糖基化酶NEIL1缺陷的小鼠表现出较高的自发突变,这种突变是由跨损伤DNA合成经过氧化诱导的碱基损伤引起的。几窝Neil1基因敲除小鼠中的动物的特点是在旷场环境中向后行走,而在笼子环境中则保持疯狂的向前运动。其他表型表现包括游泳测试失败、头部倾斜和转圈。赋予这些行为的突变图谱显示,在Ush1g基因的氨基酸 4 处引入了终止密码子。Ush1g bw/bw缺失小鼠表现出听觉和前庭缺陷,这些缺陷常见于影响内耳毛细胞功能的突变,包括完全缺乏听觉脑干反应和前庭诱发电位。与其他 Usher 综合征 I 型突变小鼠品系一样,毛细胞表型包括毛束紊乱和分裂,以及定位于第 1 行或第 2 行尖端的静纤毛蛋白质分布的改变。毛细胞的破坏和动纤毛位移表明USH1G 对于形成毛细胞动纤毛连接至关重要。与其他 Usher 1 型模型一致,与Ush1g bw /+ 对照相比, Ush1g bw/bw小鼠没有明显的视网膜变性。与之前描述的Ush1g等位基因相比,这个新的等位基因提供了该基因的第一个敲除模型。
更新日期:2023-06-16
中文翻译:
耳聋失明基因 Ush1g 的自发等位基因变异导致表型扩大
新的表型行为和特定遗传改变之间的关系通常通过目标特异性、定向诱变或化学诱变后的表型选择来发现。另一种方法是利用 DNA 修复途径的缺陷来维持遗传完整性,以应对自发引起的损伤。DNA糖基化酶NEIL1缺陷的小鼠表现出较高的自发突变,这种突变是由跨损伤DNA合成经过氧化诱导的碱基损伤引起的。几窝Neil1基因敲除小鼠中的动物的特点是在旷场环境中向后行走,而在笼子环境中则保持疯狂的向前运动。其他表型表现包括游泳测试失败、头部倾斜和转圈。赋予这些行为的突变图谱显示,在Ush1g基因的氨基酸 4 处引入了终止密码子。Ush1g bw/bw缺失小鼠表现出听觉和前庭缺陷,这些缺陷常见于影响内耳毛细胞功能的突变,包括完全缺乏听觉脑干反应和前庭诱发电位。与其他 Usher 综合征 I 型突变小鼠品系一样,毛细胞表型包括毛束紊乱和分裂,以及定位于第 1 行或第 2 行尖端的静纤毛蛋白质分布的改变。毛细胞的破坏和动纤毛位移表明USH1G 对于形成毛细胞动纤毛连接至关重要。与其他 Usher 1 型模型一致,与Ush1g bw /+ 对照相比, Ush1g bw/bw小鼠没有明显的视网膜变性。与之前描述的Ush1g等位基因相比,这个新的等位基因提供了该基因的第一个敲除模型。


























 京公网安备 11010802027423号
京公网安备 11010802027423号