Conservation Genetics Resources ( IF 1.1 ) Pub Date : 2023-07-08 , DOI: 10.1007/s12686-023-01309-3 N. Sastre , A. Mercadé , J. Casellas
|
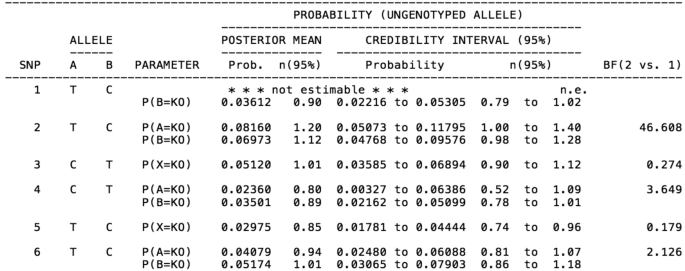
Genotyping individuals using forensic or non-invasive samples such as hair or fecal samples increases the risk of allelic amplification failure (dropout) due to the low quality and quantity of DNA. One way to decrease genotyping errors is to increase the number of replicates per sample. Here, we have developed the software SNP+ to estimate the dropout probability and the subsequent required number of replicates to obtain the reliable genotype with probability 95%. Moreover, the software predicts the minor allele frequency and compares two competing models assuming equal or allele-specific dropout probabilities by Bayes factor. The software handles data from one SNP to high density arrays (e.g., 100,000 SNPs).
中文翻译:
SNP+ 用于预测 SNP 阵列中的丢失率
使用法医或非侵入性样本(例如头发或粪便样本)对个体进行基因分型会增加由于 DNA 质量和数量较低而导致等位基因扩增失败(脱落)的风险。减少基因分型错误的一种方法是增加每个样本的重复次数。在这里,我们开发了软件 SNP+ 来估计丢失概率以及随后所需的重复次数,以获得概率为 95% 的可靠基因型。此外,该软件还可以预测次要等位基因频率,并通过贝叶斯因子假设相等或等位基因特异性丢失概率来比较两个竞争模型。该软件可处理从一个 SNP 到高密度阵列(例如 100,000 个 SNP)的数据。


























 京公网安备 11010802027423号
京公网安备 11010802027423号