Neurogenetics ( IF 2.2 ) Pub Date : 2023-07-15 , DOI: 10.1007/s10048-023-00725-9 Lianghao Si 1 , Zhanjun Wang 1 , Xu-Ying Li 1 , Yang Song 1 , Tingyan Yao 1 , Erhe Xu 1 , Xianling Wang 1 , Chaodong Wang 1
|
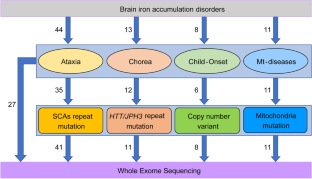
Brain iron accumulation disorders (BIADs) are a group of diseases characterized by iron overload in deep gray matter nuclei, which is a common feature of neurodegenerative diseases. Although genetic factors have been reported to be one of the etiologies, much more details about the genetic background and molecular mechanism of BIADs remain unclear. This study aimed to illustrate the genetic characteristics of BIADs and clarify their molecular mechanisms. A total of 84 patients with BIADs were recruited from April 2018 to October 2022 at Xuanwu Hospital. Clinical characteristics including family history, consanguineous marriage history, and age at onset (AAO) were collected and assessed by two senior neurologists. Neuroimaging data were conducted for all the patients, including cranial magnetic resonance imaging (MRI) and susceptibility-weighted imaging (SWI). Whole-exome sequencing (WES) and capillary electrophoresis for detecting sequence mutation and trinucleotide repeat expansion, respectively, were conducted on all patients and part of their parents (whose samples were available). Variant pathogenicity was assessed according to the American College of Medical Genetics and Association for Molecular Pathology (ACMG/AMP). The NBIA and NBIA-like genes with mutations were included for bioinformatic analysis, using Gene Ontology (GO) annotation and Kyoto Encyclopedia of Genes and Genome (KEGG). GO annotation and KEGG pathway analysis were performed on Metascape platform. In the 84 patients, 30 (35.7%) were found to carry mutations, among which 20 carried non-dynamic mutations (missense, stop-gained, frameshift, inframe, and exonic deletion) and 10 carried repeat expansion mutations. Compared with sporadic cases, familial cases had more genetic variants (non-dynamic mutation: P=0.025, dynamic mutation: P=0.003). AAO was 27.85±10.42 years in cases with non-dynamic mutations, which was significantly younger than those without mutations (43.13±17.17, t=3.724, P<0.001) and those with repeated expansions (45.40±8.90, t=4.550, P<0.001). Bioinformatic analysis suggested that genes in lipid metabolism, autophagy, mitochondria regulation, and ferroptosis pathways are more likely to be involved in the pathogenesis of BIADs. This study broadens the genetic spectrum of BIADs and has important implications in genetic counselling and clinical diagnosis. Patients diagnosed as BIADs with early AAO and family history are more likely to carry mutations. Bioinformatic analysis provides new insights into the molecular pathogenesis of BIADs, which may shed lights on the therapeutic strategy for neurodegenerative diseases.
中文翻译:
在脑铁积累障碍患者中发现的新突变和分子途径
脑铁积累障碍(BIAD)是一组以深部灰质核铁超载为特征的疾病,这是神经退行性疾病的共同特征。尽管据报道遗传因素是 BIAD 的病因之一,但有关 BIAD 遗传背景和分子机制的更多细节仍不清楚。本研究旨在阐明BIADs的遗传特征并阐明其分子机制。2018年4月至2022年10月宣武医院共招募84名BIAD患者。由两名资深神经科医生收集和评估临床特征,包括家族史、近亲结婚史和发病年龄(AAO)。对所有患者进行神经影像学数据,包括头颅磁共振成像(MRI)和磁敏感加权成像(SWI)。对所有患者及其部分父母(其样本可用)进行了全外显子组测序(WES)和毛细管电泳,分别检测序列突变和三核苷酸重复扩增。根据美国医学遗传学和分子病理学协会 (ACMG/AMP) 评估变异致病性。使用基因本体论 (GO) 注释和京都基因和基因组百科全书 (KEGG),将具有突变的 NBIA 和 NBIA 样基因纳入生物信息学分析。在Metascape平台上进行GO注释和KEGG通路分析。在84例患者中,30例(35.7%)被发现携带突变,其中20例携带非动态突变(错义、停止获得、移码、内框和外显子缺失),10例携带重复扩展突变。与散发病例相比,家族性病例有更多的遗传变异(非动态突变:P =0.025,动态突变:P =0.003)。非动态突变病例的AAO年龄为27.85±10.42岁,明显低于无突变病例(43.13±17.17,t =3.724,P <0.001)和重复扩增病例(45.40±8.90,t =4.550,P )。 <0.001)。生物信息学分析表明,脂质代谢、自噬、线粒体调节和铁死亡途径等基因更有可能参与 BIAD 的发病机制。这项研究拓宽了 BIAD 的遗传谱,对遗传咨询和临床诊断具有重要意义。诊断为 BIAD 且具有早期 AAO 和家族史的患者更有可能携带突变。生物信息学分析为 BIAD 的分子发病机制提供了新的见解,这可能有助于揭示神经退行性疾病的治疗策略。


























 京公网安备 11010802027423号
京公网安备 11010802027423号