Neurogenetics ( IF 2.2 ) Pub Date : 2023-07-19 , DOI: 10.1007/s10048-023-00726-8 Zeyu Zhu 1 , Wenzhe Hou 1, 2 , Yuwen Cao 1 , Haoran Zheng 1, 3 , Wotu Tian 1 , Li Cao 1
|
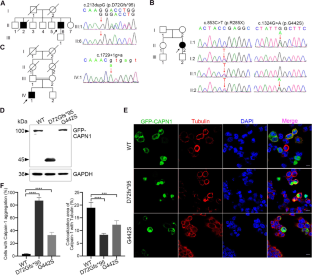
Spastic paraplegia type 76 (SPG76) is a subtype of hereditary spastic paraplegia (HSP) caused by calpain-1 (CAPN1) mutations. Our study described the phenotypic and genetic characteristics of three families with spastic ataxia due to various CAPN1 mutations and further explored the pathogenesis of the two novel mutations. The three patients were 48, 39, and 48 years old, respectively. Patients 1 and 3 were from consanguineous families, while patient 2 was sporadic. Physical examination showed hypertonia, hyperreflexia, and Babinski signs in the lower limbs. Patients 2 and 3 additionally had dysarthria and depression. CAPN1 mutations were identified by whole-exome sequencing, followed by Sanger sequencing and co-segregation analysis within the family. Functional examination of the newly identified mutations was further explored. Two homozygous mutations were detected in patient 1 (c.213dupG, p.D72Gfs*95) and patient 3 (c.1729+1G>A) with HSP, respectively. Patient 2 had compound heterozygous mutations c.853C>T (p.R285X) and c.1324G>A (p.G442S). Western blotting revealed the p.D72Gfs*95 with a smaller molecular weight than WT and p.G442S. In vitro, the wild-type calpain-1 is mostly located in the cytoplasm and colocalized with tubulin by immunostaining. However, p.D72Gfs*95 and p.G442S abnormally formed intracellular aggregation, with little colocalization with tubulin. In this study, we identified three cases with SPG76, due to four various CAPN1 mutations, presenting lower limb spasticity and ataxia, with or without bulbar involvement and emotional disorder. Among these, c.213dupG and c.1324G>A are first identified in this paper. The genotype-phenotype correlation of the SPG76 cases reported worldwide was further summarized.
中文翻译:
CAPN1新突变导致76型痉挛性截瘫:三例病例报告及文献综述
76 型痉挛性截瘫 (SPG76) 是由钙蛋白酶 1 ( CAPN1 ) 突变引起的遗传性痉挛性截瘫 (HSP) 的一个亚型。我们的研究描述了由于各种CAPN1突变而导致痉挛性共济失调的三个家系的表型和遗传特征,并进一步探讨了这两种新突变的发病机制。这三名患者年龄分别为48岁、39岁和48岁。患者1、3为近亲家庭,患者2为散发病例。查体可见下肢肌张力亢进、反射亢进、巴宾斯基征。患者 2 和 3 还患有构音障碍和抑郁症。通过全外显子组测序、随后进行桑格测序和家族内的共分离分析来鉴定CAPN1突变。进一步探索了新发现的突变的功能检查。在 HSP 患者 1 (c.213dupG, p.D72Gfs*95) 和患者 3 (c.1729+1G>A) 中分别检测到两个纯合突变。患者 2 具有复合杂合突变 c.853C>T (p.R285X) 和 c.1324G>A (p.G442S)。Western blotting显示p.D72Gfs*95的分子量比WT和p.G442S更小。在体外,免疫染色显示野生型 calpain-1 大部分位于细胞质中,并与微管蛋白共定位。然而,p.D72Gfs*95和p.G442S异常形成细胞内聚集,与微管蛋白几乎没有共定位。在这项研究中,我们发现了三例 SPG76 病例,由于四种不同的CAPN1突变,表现为下肢痉挛和共济失调,伴或不伴延髓受累和情绪障碍。其中,c.213dupG和c.1324G>A是本文首次鉴定的。进一步总结了全球报告的 SPG76 例基因型-表型相关性。


























 京公网安备 11010802027423号
京公网安备 11010802027423号