Kinetics and Catalysis ( IF 1.1 ) Pub Date : 2023-07-23 , DOI: 10.1134/s0023158423040018 P. S. Bandurist , D. A. Pichugina
|
Abstract
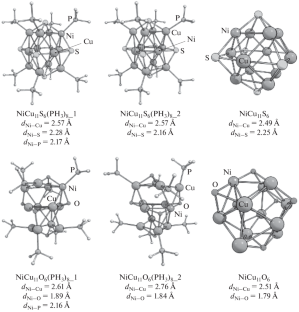
Density functional theory (DFT) (PBE) was used to simulate the breaking of C–H bond in methane on copper-enriched Ni–Cu clusters as the first step of dry reforming of methane. The models of catalysts were the nanoscale clusters NiCu11S6(PH3)8, NiCu11S6, NiCu11O6(PH3)8, and NiCu11O6. The binding energy of methane with the clusters was calculated, and the activation energy of the step CH4* → \({\text{CH}}_{3}^{*}\) + H* was determined. It was found from the obtained data that the NiCu11O6 catalytic system is the most promising for CH4 activation in terms of both kinetics (activation energy is 99 kJ/mol) and thermodynamics (energy change of the step is –29 kJ/mol). The coking resistance of the NiCu11O6 cluster was estimated by simulating the CH adsorption followed by dissociation (CH* → C* + H*). The calculated activation energy of this step is rather high: 159 kJ/mol.
中文翻译:
甲烷中 Ni-Cu 氧化物和硫化物团簇 C-H 键活化的量子化学研究
摘要
密度泛函理论 (DFT) (PBE) 用于模拟富铜 Ni-Cu 团簇上甲烷中 C-H 键的断裂,作为甲烷干重整的第一步。催化剂模型为纳米级团簇NiCu 11 S 6 (PH 3 ) 8、NiCu 11 S 6、NiCu 11 O 6 (PH 3 ) 8和NiCu 11 O 6。计算甲烷与团簇的结合能,并计算步骤CH 4 * → \({\text{CH}}_{3}^{*}\)的活化能+ H* 被测定。从获得的数据发现,NiCu 11 O 6催化体系在动力学(活化能为99 kJ/mol)和热力学(步骤能量变化为–29 kJ/mol)方面最有希望用于CH 4活化。NiCu 11 O 6团簇的抗结焦性通过模拟CH吸附随后解离(CH*→C*+H*)来估计。该步骤的计算活化能相当高:159 kJ/mol。


























 京公网安备 11010802027423号
京公网安备 11010802027423号