Interdisciplinary Sciences: Computational Life Sciences ( IF 4.8 ) Pub Date : 2023-08-21 , DOI: 10.1007/s12539-023-00584-w Jisha Augustine 1 , A S Jereesh 1
|
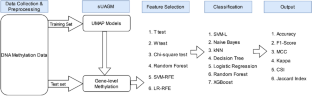
DNA methylation is an epigenetic alteration that plays a fundamental part in governing gene regulatory processes. The DNA methylation mechanism affixes methyl groups to distinct cytosine residues, influencing chromatin architectures. Multiple studies have demonstrated that DNA methylation's regulatory effect on genes is linked to the beginning and progression of several disorders. Researchers have recently uncovered thousands of phenotype-related methylation sites through the epigenome-wide association study (EWAS). However, combining the methylation levels of several sites within a gene and determining the gene-level DNA methylation remains challenging. In this study, we proposed the supervised UMAP Assisted Gene-level Methylation method (sUAGM) for disease prediction based on supervised UMAP (Uniform Manifold Approximation and Projection), a manifold learning-based method for reducing dimensionality. The methylation values at the gene level generated using the proposed method are evaluated by employing various feature selection and classification algorithms on three distinct DNA methylation datasets derived from blood samples. The performance has been assessed employing classification accuracy, F-1 score, Mathews Correlation Coefficient (MCC), Kappa, Classification Success Index (CSI) and Jaccard Index. The Support Vector Machine with the linear kernel (SVML) classifier with Recursive Feature Elimination (RFE) performs best across all three datasets. From comparative analysis, our method outperformed existing gene-level and site-level approaches by achieving 100% accuracy and F1-score with fewer genes. The functional analysis of the top 28 genes selected from the Parkinson's disease dataset revealed a significant association with the disease.
Graphical Abstract
中文翻译:
用于疾病预测的基因水平甲基化鉴定
DNA 甲基化是一种表观遗传改变,在基因调控过程中发挥着重要作用。DNA 甲基化机制将甲基固定在不同的胞嘧啶残基上,从而影响染色质结构。多项研究表明,DNA 甲基化对基因的调节作用与多种疾病的发生和进展有关。研究人员最近通过全表观基因组关联研究(EWAS)发现了数千个与表型相关的甲基化位点。然而,结合基因内多个位点的甲基化水平并确定基因水平的 DNA 甲基化仍然具有挑战性。在本研究中,我们提出了基于监督 UMAP(统一流形逼近和投影)的疾病预测监督 UMAP 辅助基因水平甲基化方法(sUAGM),这是一种基于流形学习的降维方法。通过对源自血液样本的三个不同的 DNA 甲基化数据集采用各种特征选择和分类算法来评估使用所提出的方法生成的基因水平的甲基化值。采用分类准确性、F-1 分数、Mathews 相关系数 (MCC)、Kappa、分类成功指数 (CSI) 和 Jaccard 指数来评估性能。具有线性核 (SVML) 分类器和递归特征消除 (RFE) 的支持向量机在所有三个数据集上表现最佳。通过比较分析,我们的方法通过用更少的基因实现 100% 的准确性和 F1 分数,优于现有的基因水平和位点水平方法。对从帕金森病数据集中选出的前 28 个基因进行的功能分析揭示了与该疾病的显着关联。


























 京公网安备 11010802027423号
京公网安备 11010802027423号