Neurogenetics ( IF 2.2 ) Pub Date : 2023-08-22 , DOI: 10.1007/s10048-023-00729-5 Edouard Palu 1 , Julius Järvilehto 2 , Jana Pennonen 2 , Nadine Huber 3 , Sanna-Kaisa Herukka 4, 5 , Annakaisa Haapasalo 3 , Pirjo Isohanni 2, 6 , Henna Tyynismaa 2 , Mari Auranen 7 , Emil Ylikallio 2, 7
|
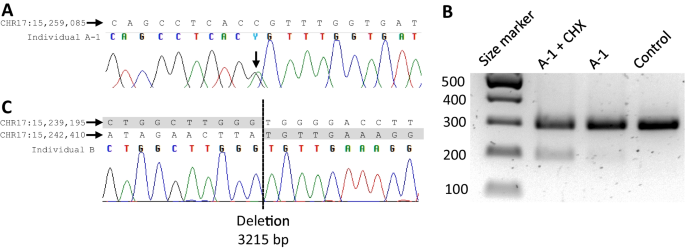
Charcot-Marie-Tooth disease (CMT) is a heterogeneous set of hereditary neuropathies whose genetic causes are not fully understood. Here, we characterize three previously unknown variants in PMP22 and assess their effect on the recently described potential CMT biomarkers’ growth differentiation factor 15 (GDF15) and neurofilament light (NFL): first, a heterozygous PMP22 c.178G > A (p.Glu60Lys) in one mother-son pair with adult-onset mild axonal neuropathy. The variant led to abnormal splicing, confirmed in fibroblasts by reverse transcription PCR. Second, a de novo PMP22 c.35A > C (p.His12Pro), and third, a heterozygous 3.2 kb deletion predicting loss of exon 4. The latter two had severe CMT and ultrasonography showing strong nerve enlargement similar to a previous case of exon 4 loss due to a larger deletion. We further studied patients with PMP22 duplication (CMT1A) finding slightly elevated plasma NFL, as measured by the single molecule array immunoassay (SIMOA). In addition, plasma GDF15, as measured by ELISA, correlated with symptom severity for CMT1A. However, in the severely affected individuals with PMP22 exon 4 deletion or p.His12Pro, these biomarkers were within the range of variability of CMT1A and controls, although they had more pronounced nerve hypertrophy. This study adds p.His12Pro and confirms PMP22 exon 4 deletion as causes of severe CMT, whereas the previously unknown splice variant p.Glu60Lys leads to mild axonal neuropathy. Our results suggest that GDF15 and NFL do not distinguish CMT1A from advanced hypertrophic neuropathy caused by rare PMP22 variants.
中文翻译:
轻度至重度神经病变中罕见的 PMP22 变异与血浆 GDF15 或神经丝光不相关
腓骨肌萎缩症 (CMT) 是一组异质性遗传性神经病,其遗传原因尚不完全清楚。在这里,我们描述了PMP22中三个以前未知的变异,并评估了它们对最近描述的潜在 CMT 生物标志物生长分化因子 15 (GDF15) 和神经丝光 (NFL) 的影响:首先,杂合PMP22 c.178G > A (p.Glu60Lys )一对患有成人发病的轻度轴突神经病的母子。该变异导致异常剪接,通过逆转录 PCR 在成纤维细胞中得到证实。其次,从头开始PMP22 c.35A > C (p.His12Pro),第三,预测外显子 4 丢失的杂合 3.2 kb 缺失。后两者具有严重的 CMT 和超声检查,显示与之前的外显子病例类似的强烈神经增大4.由于较大的删除而造成的损失。我们进一步研究了PMP22重复 (CMT1A)患者,通过单分子阵列免疫分析 (SIMOA) 测量,发现血浆 NFL 略有升高。此外,通过 ELISA 测量的血浆 GDF15 与 CMT1A 症状严重程度相关。然而,在PMP22外显子 4 缺失或 p.His12Pro严重受影响的个体中,这些生物标志物在 CMT1A 和对照的变异范围内,尽管他们有更明显的神经肥大。这项研究添加了 p.His12Pro 并证实PMP22外显子 4 缺失是严重 CMT 的原因,而先前未知的剪接变异 p.Glu60Lys 会导致轻度轴突神经病变。我们的结果表明,GDF15 和 NFL 不能区分 CMT1A 和罕见PMP22变异引起的晚期肥厚性神经病。


























 京公网安备 11010802027423号
京公网安备 11010802027423号