Journal of Combinatorial Optimization ( IF 1 ) Pub Date : 2023-09-10 , DOI: 10.1007/s10878-023-01083-w Gabriel Siqueira , Alexsandro Oliveira Alexandrino , Zanoni Dias
|
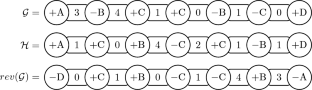
Genome rearrangement distance problems allow to estimate the evolutionary distance between genomes. These problems aim to compute the minimum number of mutations called rearrangement events necessary to transform one genome into another. Two commonly studied rearrangements are the reversal, which inverts a sequence of genes, and the transposition, which exchanges two consecutive sequences of genes. Seminal works on this topic focused on the sequence of genes and assumed that each gene occurs exactly once on each genome. More realistic models have been assuming that a gene may have multiple copies or may appear in only one of the genomes. Other models also take into account the nucleotides between consecutive pairs of genes, which are called intergenic regions. This work combines all these generalizations defining the signed intergenic reversal distance (SIRD), the signed intergenic reversal and transposition distance (SIRTD), the signed intergenic reversal and indels distance (SIRID), and the signed intergenic reversal, transposition, and indels distance (SIRTID) problems. We show a relation between these problems and the signed minimum common intergenic string partition (SMCISP) problem. From such relation, we derive \(\varTheta (k)\)-approximation algorithms for the SIRD and the SIRTD problems, where k is maximum number of copies of a gene in the genomes. These algorithms also work as heuristics for the SIRID and SIRTID problems. Additionally, we present some parametrized algorithms for SMCISP that ensure constant approximation factors for the distance problems. Our experimental tests on simulated genomes show an improvement on the rearrangement distances with the use of the partition algorithms.
中文翻译:
考虑重复基因、基因间区域和插入缺失的有符号重排距离
基因组重排距离问题可以估计基因组之间的进化距离。这些问题旨在计算将一个基因组转变为另一个基因组所需的最小突变数(称为重排事件)。两种经常研究的重排是反转(反转基因序列)和转座(交换两个连续的基因序列)。该主题的开创性工作主要关注基因序列,并假设每个基因在每个基因组上恰好出现一次。更现实的模型假设一个基因可能有多个拷贝,或者可能只出现在一个基因组中。其他模型还考虑了连续基因对之间的核苷酸,称为基因间区域。这项工作结合了定义有符号基因间反转距离 (SIRD)、有符号基因间反转和转座距离 (SIRTD)、有符号基因间反转和插入缺失距离 (SIRID) 以及有符号基因间反转、转座和插入缺失距离的所有这些概括( SIRTID)问题。我们展示了这些问题与有符号最小公共基因间字符串划分(SMCISP)问题之间的关系。从这样的关系,我们得出\(\varTheta (k)\) - SIRD 和 SIRTD 问题的近似算法,其中k是基因组中基因的最大拷贝数。这些算法还可以作为 SIRID 和 SIRTID 问题的启发式算法。此外,我们还提出了一些 SMCISP 参数化算法,确保距离问题的近似因子恒定。我们对模拟基因组的实验测试表明,使用分区算法可以改善重排距离。


























 京公网安备 11010802027423号
京公网安备 11010802027423号