Journal of Structural Biology ( IF 3 ) Pub Date : 2023-09-25 , DOI: 10.1016/j.jsb.2023.108030 E Fernandez-Gimenez 1 , J M Carazo 2 , C O S Sorzano 2
|
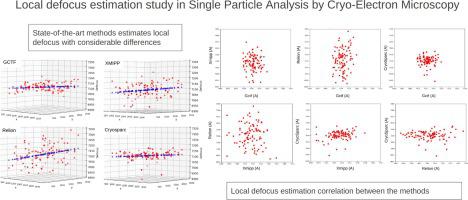
Single Particle analysis (SPA) aims to determine the three-dimensional structure of proteins and macromolecular complexes. The current state of the art has allowed us to achieve near-atomic and even atomic resolutions. To obtain high-resolution structures, a set of well-defined image processing steps is required. A critical one is the estimation of the Contrast Transfer Function (CTF), which considers the sample defocus and aberrations of the microscope. Defocus is usually globally estimated; in this case, it is the same for all the particles in each micrograph. But proteins are ice-embedded at different heights, suggesting that defocus should be measured in a local (per particle) manner. There are four state-of-the-art programs to estimate local defocus (Gctf, Relion, CryoSPARC, and Xmipp). In this work, we have compared the results of these software packages to check whether the resolution improves. We have used the Scipion framework and developed a specific program to analyze local defocus. The results produced by different programs do not show a clear consensus using the current test datasets in this study.
中文翻译:
冷冻电子显微镜单粒子分析中的局部散焦估计
单粒子分析(SPA)旨在确定蛋白质和大分子复合物的三维结构。目前的技术水平使我们能够实现近原子甚至原子分辨率。为了获得高分辨率结构,需要一组明确的图像处理步骤。其中一个关键是对比度传递函数(CTF)的估计,它考虑了样本散焦和显微镜的像差。散焦通常是全局估计的;在这种情况下,每张显微照片中的所有颗粒都是相同的。但蛋白质以不同的高度嵌入冰中,这表明应该以局部(每个粒子)的方式测量散焦。有四种最先进的程序可用于估计局部散焦(Gctf、Relion、CryoSPARC 和 Xmipp)。在这项工作中,我们比较了这些软件包的结果,以检查分辨率是否有所提高。我们使用Scipion框架并开发了一个具体的程序来分析局部散焦。使用本研究中当前的测试数据集,不同程序产生的结果并未显示出明确的共识。


























 京公网安备 11010802027423号
京公网安备 11010802027423号