Medical Microbiology and Immunology ( IF 5.4 ) Pub Date : 2023-10-04 , DOI: 10.1007/s00430-023-00783-8 Rasha Emad 1 , Iman S Naga 2
|
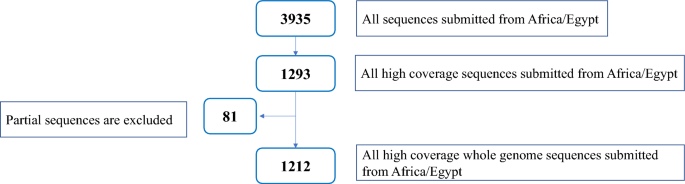
Several tools have been developed for severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) genotyping based on either whole genome or spike sequencing. We aimed to highlight the molecular epidemiological landscape of SARS-CoV-2 in Egypt since the start of the pandemic, to describe discrepancies between the 3 typing tools: Global Initiative on Sharing Avian Influenza Data (GISAID), Nextclade, and Phylogenetic Assignment of Named Global Outbreak Lineages (PANGOLIN) and to assess the fitness of spike and nucleocapsid regions for lineage assignment compared to the whole genome. A total of 3935 sequences isolated from Egypt (March 2020–2023) were retrieved from the GISAID database. A subset of data (n = 1212) with high coverage whole genome was used for tool discrimination and agreement analyses. Among 1212 sequences, the highest discriminatory power was 0.895 for PANGOLIN, followed by GISAID (0.872) and Nextclade (0.866). There was a statistically significant difference (p = 0.0418) between lineages assigned via spike (30%) and nucleocapsid (46%) compared to their whole genome-assigned lineages. The first 3 pandemic waves were dominated by B.1, followed by C.36 and then C.36.3, while the fourth to sixth waves were dominated by the B.1.617.2, BA, and BA.5.2 lineages, respectively. Current shift in lineage typing to recombinant forms. The 3 typing tools showed comparable discrimination among SARS-CoV-2 lineages. The nucleocapsid region could be used for lineage assignment.
中文翻译:
埃及患者中 SARS-CoV-2 的比较基因分型:近全长基因组序列与选定的刺突和核衣壳区域
已经开发了多种基于全基因组或尖峰测序的严重急性呼吸综合征冠状病毒-2 (SARS-CoV-2) 基因分型工具。我们的目的是强调自大流行开始以来埃及 SARS-CoV-2 的分子流行病学状况,描述 3 种分型工具之间的差异:全球禽流感数据共享倡议 (GISAID)、Nextclade 和命名系统发育分配全球爆发谱系 (PANGOLIN) 并评估刺突和核衣壳区域与整个基因组相比的谱系分配的适应性。从 GISAID 数据库中检索到从埃及(2020 年 3 月至 2023 年)分离的总共 3935 个序列。具有高覆盖率全基因组的数据子集(n = 1212)用于工具辨别和一致性分析。在 1212 个序列中,PANGOLIN 的判别力最高,为 0.895,其次是 GISAID (0.872) 和 Nextclade (0.866)。与整个基因组分配的谱系相比,通过尖峰(30%)和核衣壳(46%)分配的谱系之间存在统计学显着差异(p = 0.0418)。前 3 波大流行以 B.1 为主,其次是 C.36,然后是 C.36.3,而第四至第六波分别以 B.1.617.2、BA 和 BA.5.2 谱系为主。当前谱系分型向重组形式的转变。这 3 种分型工具在 SARS-CoV-2 谱系中表现出类似的区分能力。核衣壳区域可用于谱系分配。


























 京公网安备 11010802027423号
京公网安备 11010802027423号