Journal of Computer-Aided Molecular Design ( IF 3.5 ) Pub Date : 2023-10-25 , DOI: 10.1007/s10822-023-00538-w Sadra Kashef Ol Gheta 1 , Anne Bonin 1 , Thomas Gerlach 2, 3 , Andreas H Göller 1
|
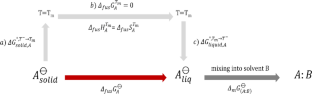
In this study, we use machine learning algorithms with QM-derived COSMO-RS descriptors, along with Morgan fingerprints, to predict the absolute solubility of drug-like compounds. The QM-derived descriptors account for the molecular properties of the solute, i.e., the solute–solute interactions in an artificial-liquid-state (super-cooled liquid), and the solute–solvent interactions in solution. We employ two main approaches to predict solubility: (i) a hypothetical pathway that involves melting the solute at room temperature T = T¯ (\({\Delta }_{fus}{G}_{A}^{\ominus }\)) and mixing the artificially liquid solute into the solvent (\({\Delta }_{m}{G}_{\left(A:B\right)}^{\ominus }\)). In this approach \({\Delta }_{fus}{G}_{A}^{\ominus }\) is predicted using machine learning models, and the \({\Delta }_{m}{G}_{\left(A:B\right)}^{\ominus }\) is obtained from COSMO-RS calculations; (ii) direct solubility prediction using machine learning algorithms. The models were trained on a large number of Bayer in-house compounds for which water solubility data is available at physiological pH of 6.5 and ambient temperature. We also evaluated our models using external datasets from a solubility challenge. Our models present great improvements compared to the absolute solubility prediction with the QSAR model for the artificial liquid state as implemented in the COSMOtherm software, for both in-house and external datasets. We are furthermore able to demonstrate the superiority of QM-derived descriptors compared to cheminformatics descriptors. We finally present low-cost alternative models using fragment-based COSMOquick calculations with only marginal reduction in the quality of predicted solubility.
中文翻译:
通过应用机器学习模型以人工液态作为固态的代理来预测绝对水溶性
在这项研究中,我们使用机器学习算法和 QM 衍生的 COSMO-RS 描述符以及摩根指纹来预测类药物化合物的绝对溶解度。QM 导出的描述符解释了溶质的分子特性,即人工液体状态(过冷液体)中的溶质-溶质相互作用,以及溶液中的溶质-溶剂相互作用。我们采用两种主要方法来预测溶解度:(i)一种假设途径,涉及在室温下熔化溶质 T = T¯ ( \({\Delta }_{fus}{G}_{A}^{\ominus } \))并将人工液体溶质混合到溶剂中(\({\Delta }_{m}{G}_{\left(A:B\right)}^{\ominus }\) )。在这种方法中,使用机器学习模型来预测\({\Delta }_{fus}{G}_{A}^{\ominus }\),并且\({\Delta }_{m}{G}_ {\left(A:B\right)}^{\ominus }\)由 COSMO-RS 计算得到;(ii) 使用机器学习算法直接预测溶解度。这些模型针对大量拜耳内部化合物进行了训练,这些化合物在生理 pH 值 6.5 和环境温度下可获得水溶性数据。我们还使用来自溶解度挑战的外部数据集评估了我们的模型。与 COSMO therm软件中实施的人工液态 QSAR 模型的绝对溶解度预测相比,我们的模型在内部和外部数据集上都有很大的改进。此外,我们还能够证明 QM 衍生描述符相对于化学信息学描述符的优越性。我们最终提出了使用基于碎片的 COSMO快速计算的低成本替代模型,仅略微降低了预测溶解度的质量。


























 京公网安备 11010802027423号
京公网安备 11010802027423号