当前位置:
X-MOL 学术
›
Genes Cells
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
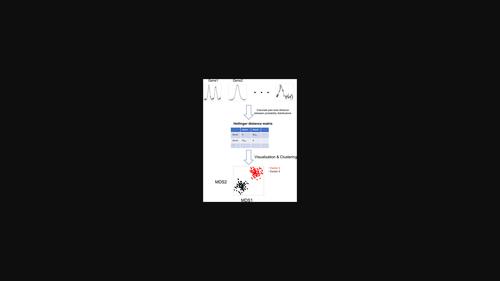
Visualization of the landscape of the read alignment shape of ATAC-seq data using Hellinger distance metric
Genes to Cells ( IF 2.1 ) Pub Date : 2023-11-21 , DOI: 10.1111/gtc.13082 Jian Hao Cheng 1 , Cheng Zheng 1 , Ryo Yamada 1 , Daigo Okada 1
Genes to Cells ( IF 2.1 ) Pub Date : 2023-11-21 , DOI: 10.1111/gtc.13082 Jian Hao Cheng 1 , Cheng Zheng 1 , Ryo Yamada 1 , Daigo Okada 1
Affiliation
|
Assay for Transposase-Accessible Chromatin using high-throughput sequencing (ATAC-seq) is the popular technique using next-generation sequencing to measure chromatin accessibility and identify open chromatin regions. While read alignment shape information of next-generation sequencing data with intensity information has been used in various bioinformatics methods, few studies have focused on pure shape information alone. In this study, we investigated what types of ATAC-seq read alignment shapes are observed for the promoter region and whether the pure shape information was related or unrelated to other gene features. We introduced a novel concept and pipeline for handling the pure shape information of NGS data as probability distributions and quantifying their dissimilarities by information theory. Based on this concept, we demonstrate that the pure shape information of ATAC-seq data is correlated with chromatin openness and some gene characteristics. On the other hand, it is suggested that the pure information of ATAC-seq read alignment shape is unlikely to contain additional information to explain differences in RNA expression. Our study suggests that viewing the read alignment shape of NGS data as probability distributions enables us to capture the characteristics of the genome-wide landscape of such data in a non-parametric manner.
中文翻译:

使用 Hellinger 距离度量对 ATAC-seq 数据的读取对齐形状进行可视化
使用高通量测序 (ATAC-seq) 检测转座酶可及染色质是一种流行的技术,使用新一代测序来测量染色质可及性并识别开放染色质区域。虽然具有强度信息的下一代测序数据的读取比对形状信息已被用于各种生物信息学方法中,但很少有研究单独关注纯形状信息。在本研究中,我们研究了在启动子区域观察到什么类型的 ATAC-seq 读段比对形状,以及纯形状信息是否与其他基因特征相关或无关。我们引入了一种新颖的概念和流程,用于将 NGS 数据的纯形状信息处理为概率分布,并通过信息论量化它们的差异。基于这个概念,我们证明了 ATAC-seq 数据的纯形状信息与染色质开放性和一些基因特征相关。另一方面,表明 ATAC-seq 读段比对形状的纯信息不太可能包含解释 RNA 表达差异的附加信息。我们的研究表明,将 NGS 数据的读取比对形状视为概率分布使我们能够以非参数方式捕获此类数据的全基因组景观的特征。
更新日期:2023-11-21
中文翻译:
使用 Hellinger 距离度量对 ATAC-seq 数据的读取对齐形状进行可视化
使用高通量测序 (ATAC-seq) 检测转座酶可及染色质是一种流行的技术,使用新一代测序来测量染色质可及性并识别开放染色质区域。虽然具有强度信息的下一代测序数据的读取比对形状信息已被用于各种生物信息学方法中,但很少有研究单独关注纯形状信息。在本研究中,我们研究了在启动子区域观察到什么类型的 ATAC-seq 读段比对形状,以及纯形状信息是否与其他基因特征相关或无关。我们引入了一种新颖的概念和流程,用于将 NGS 数据的纯形状信息处理为概率分布,并通过信息论量化它们的差异。基于这个概念,我们证明了 ATAC-seq 数据的纯形状信息与染色质开放性和一些基因特征相关。另一方面,表明 ATAC-seq 读段比对形状的纯信息不太可能包含解释 RNA 表达差异的附加信息。我们的研究表明,将 NGS 数据的读取比对形状视为概率分布使我们能够以非参数方式捕获此类数据的全基因组景观的特征。


























 京公网安备 11010802027423号
京公网安备 11010802027423号