Journal of Computer-Aided Molecular Design ( IF 3.5 ) Pub Date : 2023-12-16 , DOI: 10.1007/s10822-023-00541-1 Aleksei Kabedev , Christel A. S. Bergström , Per Larsson
|
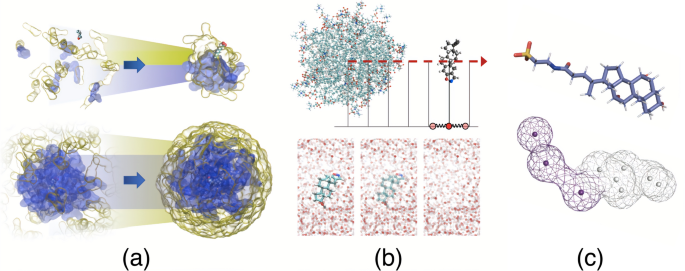
Theoretical predictions of the solubilizing capacity of micelles and vesicles present in intestinal fluid are important for the development of new delivery techniques and bioavailability improvement. A balance between accuracy and computational cost is a key factor for an extensive study of numerous compounds in diverse environments. In this study, we aimed to determine an optimal molecular dynamics (MD) protocol to evaluate small-molecule interactions with micelles composed of bile salts and phospholipids. MD simulations were used to produce free energy profiles for three drug molecules (danazol, probucol, and prednisolone) and one surfactant molecule (sodium caprate) as a function of the distance from the colloid center of mass. To address the challenges associated with such tasks, we compared different simulation setups, including freely assembled colloids versus pre-organized spherical micelles, full free energy profiles versus only a few points of interest, and a coarse-grained model versus an all-atom model. Our findings demonstrate that combining these techniques is advantageous for achieving optimal performance and accuracy when evaluating the solubilization capacity of micelles.
Graphical abstract
All-atom (AA) and coarse-grained (CG) umbrella sampling (US) simulations and point-wise free energy (FE) calculations were compared to their efficiency to computationally analyze the solubilization of active pharmaceutical ingredients in intestinal fluid colloids.
中文翻译:
胶束-小分子相互作用的分子动力学研究:制定广泛比较的策略
肠液中胶束和囊泡溶解能力的理论预测对于开发新的递送技术和提高生物利用度非常重要。准确性和计算成本之间的平衡是在不同环境中广泛研究多种化合物的关键因素。在这项研究中,我们的目的是确定最佳的分子动力学 (MD) 方案来评估小分子与胆汁盐和磷脂组成的胶束的相互作用。 MD 模拟用于生成三种药物分子(达那唑、普罗布考和泼尼松龙)和一种表面活性剂分子(癸酸钠)的自由能曲线,作为距胶体质心距离的函数。为了解决与此类任务相关的挑战,我们比较了不同的模拟设置,包括自由组装的胶体与预组织的球形胶束、完整的自由能分布与仅几个感兴趣点以及粗粒度模型与全原子模型。我们的研究结果表明,在评估胶束的增溶能力时,结合这些技术有利于实现最佳性能和准确性。
图形概要
将全原子 (AA) 和粗粒度 (CG) 伞式采样 (US) 模拟和逐点自由能 (FE) 计算与其效率进行比较,以计算分析活性药物成分在肠液胶体中的溶解情况。


























 京公网安备 11010802027423号
京公网安备 11010802027423号