Bulletin of Materials Science ( IF 1.8 ) Pub Date : 2024-01-12 , DOI: 10.1007/s12034-023-03088-x Muhammad Riaz , Syed Mansoor Ali , H Kassim , Mubasher Ali
|
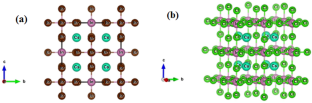
Halide perovskite materials belong to a fascinating novel class, and recently captured incredible attention as a leading aspirant for next-generation photovoltaic technology, because of their flexible chemistry and extraordinary optoelectronic properties, they also deliver a pattern for designing new materials for energy conversion and energy storage applications. In this study, we thoroughly explore the structural, electronic, optical and mechanical properties of the inorganic halide cubic perovskites (CsInX3; X = Br, Cl) for solar cell (SC) application, simulated under density functional theory (DFT) based WIEN2k code with PBE-GGA approximation. The optimized lattice parameters were found to be 11.67 Ǻ for CsInBr3 and 11.16 Ǻ for CsInCl3, respectively. The variation in the structural parameters with changing the halogen atom from Br to Cl was observed. The electronic and optical properties were computed by TB-mBJ method. These halide perovskites reveal an indirect energy bandgap quantified as 1.22 and 2.2 eV, high absorption coefficient and low reflectivity. The elastic constants (C11, C12 and C44) followed the Born stability condition, and confirmed the mechanical stability. According to the Poisson ratio (ν) and Pugh’s ratio (B/G), materials endorsed ductile behaviour as well exhibit anisotropic nature. Moreover, our theoretical findings suggested that the investigated materials have high absorption coefficients, high conductivity and low reflectivity, which makes them promising contender for SC applications.
中文翻译:
使用 DFT 研究卤化物钙钛矿 (CsInX3; X = Br, Cl) 的结构、光电和机械性能作为钙钛矿太阳能电池的吸收层
卤化物钙钛矿材料属于一类令人着迷的新颖材料,最近作为下一代光伏技术的领先者而引起了极大的关注,因为它们具有灵活的化学性质和非凡的光电性能,它们还为设计用于能源转换和能源的新材料提供了一种模式存储应用。在这项研究中,我们深入探索了用于太阳能电池(SC)应用的无机卤化物立方钙钛矿(CsInX 3 ; X = Br, Cl)的结构、电子、光学和机械性能,并在基于 WIEN2k 的密度泛函理论(DFT)下进行了模拟使用 PBE-GGA 近似的代码。结果发现,CsInBr 3 的优化晶格参数为 11.67 ε ,CsInCl 3的优化晶格参数分别为 11.16 ε 。观察到结构参数随着卤素原子从 Br 变为 Cl 的变化。通过TB-mBJ方法计算电子和光学性质。这些卤化物钙钛矿具有量化为 1.22 和 2.2 eV 的间接能带隙、高吸收系数和低反射率。弹性常数(C 11、C 12和C 44 )遵循Born稳定条件,并证实了机械稳定性。根据泊松比 (ν) 和皮尤比 ( B / G ),材料不仅具有延展性,而且表现出各向异性。此外,我们的理论研究结果表明,所研究的材料具有高吸收系数、高电导率和低反射率,这使得它们成为 SC 应用的有希望的竞争者。


























 京公网安备 11010802027423号
京公网安备 11010802027423号