Virchows Archiv ( IF 3.5 ) Pub Date : 2024-01-20 , DOI: 10.1007/s00428-023-03730-3 Konstantin Bräutigam , Cédric Nesti , Philipp Riss , Christian Scheuba , Bruno Niederle , Tobias Grob , Annunziata Di Domenico , Maja Neuenschwander , Peter Mazal , Nastassja Köhn , Roman Trepp , Aurel Perren , Reto M. Kaderli
|
Primary hyperparathyroidism with parathyroid tumors is a typical manifestation of Multiple Endocrine Neoplasia Type 1 (MEN1) and is historically termed “primary hyperplasia”. Whether these tumors represent a multi-glandular clonal disease or hyperplasia has not been robustly proven so far. Loss of Menin protein expression is associated with inactivation of both alleles and a good surrogate for a MEN1 gene mutation. The cyclin-dependent kinase inhibitor 1B (CDKN1B) gene is mutated in MEN4 and encodes for protein p27 whose expression is poorly studied in the syndromic MEN1 setting.
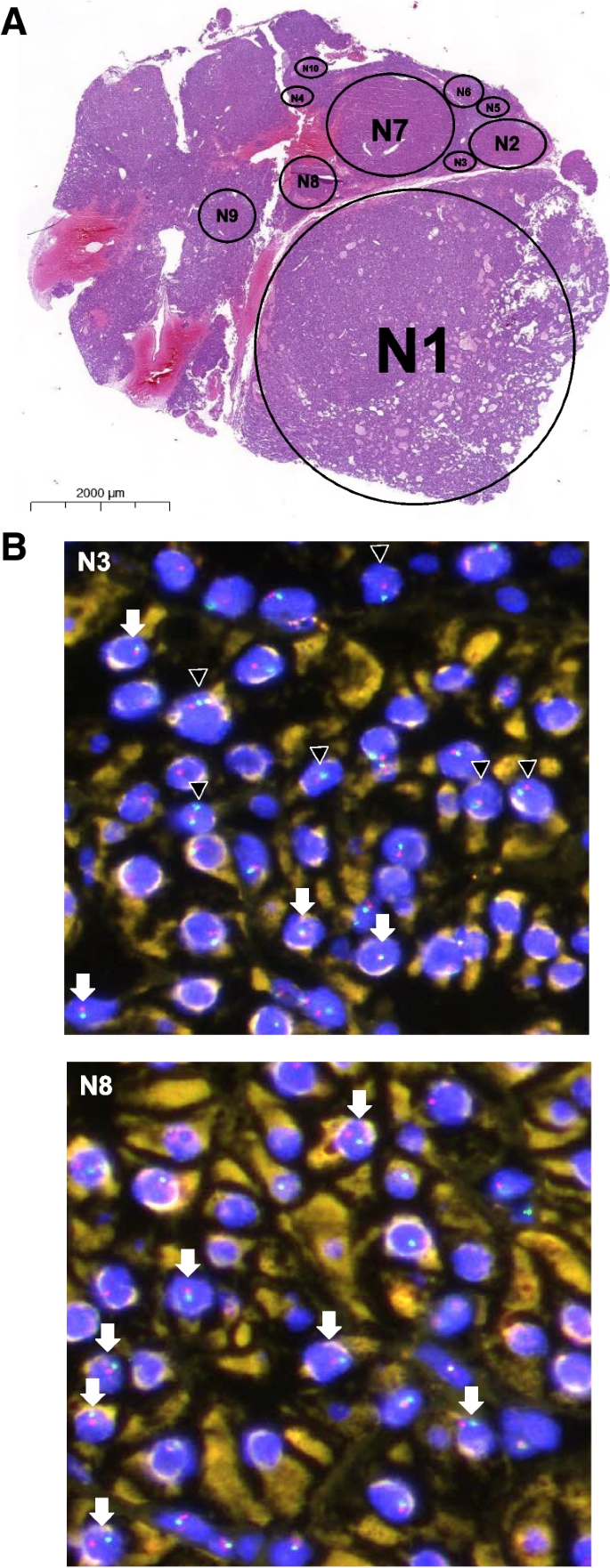
Here, we analyzed histomorphology and protein expression of Menin and p27 in parathyroid adenomas of 25 patients of two independent, well-characterized MEN1 cohorts. The pattern of loss of heterozygosity (LOH) was assessed by fluorescence in situ hybridization (FISH) in one MEN1-associated parathyroid adenoma. Further, next-generation sequencing (NGS) was performed on eleven nodules of four MEN1 patients.
Morphologically, the majority of MEN1 adenomas consisted of multiple distinct nodules, in which Menin expression was mostly lost and p27 protein expression reduced. FISH analysis revealed that most nodules exhibited MEN1 loss, with or without the loss of centromere 11. NGS demonstrated both subclonal evolution and the existence of clonally unrelated tumors.
Syndromic MEN1 parathyroid adenomas therefore consist of multiple clones with subclones, which supports the current concept of the novel WHO classification of parathyroid tumors (2022). p27 expression was lost in a large fraction of MEN1 parathyroids and must therefore be used with caution in suggesting MEN4.
中文翻译:
综合征型 MEN1 甲状旁腺腺瘤由亚克隆结节和克隆独立肿瘤组成
原发性甲状旁腺功能亢进症合并甲状旁腺肿瘤是多发性内分泌肿瘤1型(MEN1)的典型表现,历史上被称为“原发性增生”。迄今为止,这些肿瘤是否代表多腺体克隆性疾病或增生尚未得到有力证明。Menin 蛋白表达的丧失与两个等位基因的失活有关,并且是MEN1基因突变的良好替代物。细胞周期蛋白依赖性激酶抑制剂 1B ( CDKN1B ) 基因在 MEN4 中发生突变,编码蛋白质 p27,其表达在综合征 MEN1 环境中的研究很少。
在这里,我们分析了两个独立的、特征良好的 MEN1 队列的 25 名患者的甲状旁腺腺瘤中 Menin 和 p27 的组织形态学和蛋白表达。通过荧光原位杂交 (FISH) 评估一种 MEN1 相关甲状旁腺腺瘤的杂合性丢失 (LOH) 模式。此外,对 4 名 MEN1 患者的 11 个结节进行了下一代测序 (NGS)。
形态学上,大多数MEN1腺瘤由多个不同的结节组成,其中Menin表达大部分缺失,p27蛋白表达减少。FISH 分析显示,大多数结节表现出MEN1缺失,伴或不伴着丝粒 11 缺失。NGS 证明了亚克隆进化和克隆无关肿瘤的存在。
因此,综合征型 MEN1 甲状旁腺腺瘤由多个克隆和亚克隆组成,这支持了 WHO 甲状旁腺肿瘤新分类 (2022) 的当前概念。p27 表达在大部分 MEN1 甲状旁腺中丢失,因此在提示 MEN4 时必须谨慎使用。


























 京公网安备 11010802027423号
京公网安备 11010802027423号