当前位置:
X-MOL 学术
›
J. Photochem. Photobiol. A Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Test of the orbital-based LI3 index as a predictor of the height of the 3MLCT → 3MC transition-state barrier for gas-phase [Ru(N∧N)3]2+ polypyridine complexes
Journal of Photochemistry and Photobiology A: Chemistry ( IF 4.3 ) Pub Date : 2024-02-02 , DOI: 10.1016/j.jphotochem.2024.115502 Denis Magero , Ala Aldin M.H.M. Darghouth , Mark E. Casida
Journal of Photochemistry and Photobiology A: Chemistry ( IF 4.3 ) Pub Date : 2024-02-02 , DOI: 10.1016/j.jphotochem.2024.115502 Denis Magero , Ala Aldin M.H.M. Darghouth , Mark E. Casida
|
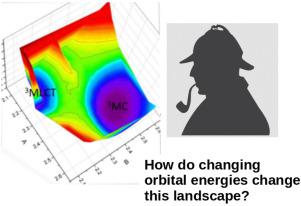
Ruthenium(II) polypyridine compounds often luminesce, with ligand-dependant lifetimes. This is the third in a series of papers devoted to finding orbital-based luminescence indices (LI) for predicting luminescence lifetimes. The third attempt (LI3) was based upon a frontier molecular orbital (FMO) like estimate of the height of the barrier to conversion from an initial phosophorescent triplet metal-ligand charge transfer (MLCT) state to a nonluminescent triplet metal-centered (MC) state which decays nonradiatively. A linear correlation was found between LI3 and extracted empirical average MLCT MC transition state (TS) barrier heights (s) which were believed to be quantitative but which were believed to capture the trends in the true barrier heights correctly. As it is known that is a large understimate of the true MLCT MC TS barrier height in the case of the bipyridine ruthenium(II) cation { [Ru(bpy)] }, but accurate TS barrier heights are difficult to obtain experimentally, it was judged useful to verify the ideas used to derive the LI3 index in the present work by calculating the energetics of the gas-phase MLCT MC reaction for a series of ruthenium(II) bipyridine complexes using the same density functional and basis sets used in calculating LI3. Specifically, four closely-related bipyridine complexes { [Ru(NN)] with NN = bpy (), 4,4’-dm-bpy (), 4,4’-dph-bpy (), and 4,4’-DTB-bpy () } were used for these calculations. In the process, we examined the gas-phase dissociation mechanism in greater detail than has been previously done and uncovered a two part mechanism. In the first part, the electron is transferred to a single ligand rather than symmetrically to all three ligands. It is the two Ru-N bonds to this ligand which are equally elongated in the transition state. The intrinsic reaction coordinate then continues down a ridge in hyperspace and bifurcates into one of two symmetry-equivalent MC structures with elongated bonds. Interestingly, no significant difference is found for the TS barriers for the four complexes treated here. Instead, LI3 is linearly correlated with the energy difference , different from what was originally intended but still consistent with the FMO arguments underlying the derivation of LI3.
中文翻译:

测试基于轨道的 LI3 指数作为气相 [Ru(N∧N)3]2+ 聚吡啶配合物 3MLCT → 3MC 过渡态势垒高度的预测因子
钌(II)聚吡啶化合物通常会发光,其寿命取决于配体。这是致力于寻找基于轨道的发光指数(LI)以预测发光寿命的系列论文中的第三篇。第三次尝试(LI3)是基于前沿分子轨道(FMO)对从初始磷光三重态金属配体电荷转移(MLCT)状态到不发光三重态金属中心(MC)转换的势垒高度的估计。非辐射衰变的状态。在 LI3 和提取的经验平均 MLCT MC 过渡态 (TS) 势垒高度 (s) 之间发现了线性相关性,该高度被认为是定量的,但被认为正确捕获了真实势垒高度的趋势。众所周知,在联吡啶钌(II)阳离子{[Ru(bpy)]}的情况下,这大大低估了真实的MLCT MC TS势垒高度,但准确的TS势垒高度很难通过实验获得,因此通过使用与计算 LI3 相同的密度泛函和基组来计算一系列钌 (II) 联吡啶络合物的气相 MLCT MC 反应的能量,被认为有助于验证当前工作中用于推导 LI3 指数的想法。具体来说,四个密切相关的联吡啶配合物{[Ru(NN)],其中 NN = bpy ()、4,4'-dm-bpy ()、4,4'-dph-bpy () 和 4,4'- DTB-bpy () } 用于这些计算。在此过程中,我们比以前更详细地研究了气相解离机制,并揭示了两部分机制。在第一部分中,电子转移到单个配体,而不是对称地转移到所有三个配体。该配体的两个 Ru-N 键在过渡态下同样伸长。然后,内在反应坐标继续沿着超空间中的脊向下延伸,并分叉成两个具有拉长键的对称等效 MC 结构之一。有趣的是,这里处理的四种复合物的 TS 屏障没有发现显着差异。相反,LI3 与能量差 线性相关,这与最初的预期不同,但仍然与 LI3 推导背后的 FMO 论点一致。
更新日期:2024-02-02
中文翻译:
测试基于轨道的 LI3 指数作为气相 [Ru(N∧N)3]2+ 聚吡啶配合物 3MLCT → 3MC 过渡态势垒高度的预测因子
钌(II)聚吡啶化合物通常会发光,其寿命取决于配体。这是致力于寻找基于轨道的发光指数(LI)以预测发光寿命的系列论文中的第三篇。第三次尝试(LI3)是基于前沿分子轨道(FMO)对从初始磷光三重态金属配体电荷转移(MLCT)状态到不发光三重态金属中心(MC)转换的势垒高度的估计。非辐射衰变的状态。在 LI3 和提取的经验平均 MLCT MC 过渡态 (TS) 势垒高度 (s) 之间发现了线性相关性,该高度被认为是定量的,但被认为正确捕获了真实势垒高度的趋势。众所周知,在联吡啶钌(II)阳离子{[Ru(bpy)]}的情况下,这大大低估了真实的MLCT MC TS势垒高度,但准确的TS势垒高度很难通过实验获得,因此通过使用与计算 LI3 相同的密度泛函和基组来计算一系列钌 (II) 联吡啶络合物的气相 MLCT MC 反应的能量,被认为有助于验证当前工作中用于推导 LI3 指数的想法。具体来说,四个密切相关的联吡啶配合物{[Ru(NN)],其中 NN = bpy ()、4,4'-dm-bpy ()、4,4'-dph-bpy () 和 4,4'- DTB-bpy () } 用于这些计算。在此过程中,我们比以前更详细地研究了气相解离机制,并揭示了两部分机制。在第一部分中,电子转移到单个配体,而不是对称地转移到所有三个配体。该配体的两个 Ru-N 键在过渡态下同样伸长。然后,内在反应坐标继续沿着超空间中的脊向下延伸,并分叉成两个具有拉长键的对称等效 MC 结构之一。有趣的是,这里处理的四种复合物的 TS 屏障没有发现显着差异。相反,LI3 与能量差 线性相关,这与最初的预期不同,但仍然与 LI3 推导背后的 FMO 论点一致。


























 京公网安备 11010802027423号
京公网安备 11010802027423号