Metabolic Brain Disease ( IF 3.6 ) Pub Date : 2024-02-16 , DOI: 10.1007/s11011-024-01343-6 Marwa Maalej , Lamia Sfaihi , Olfa-Alila Fersi , Boudour Khabou , Marwa Ammar , Rahma Felhi , Marwa Kharrat , Jihen Chouchen , Thouraya Kammoun , Abdelaziz Tlili , Faiza Fakhfakh
|
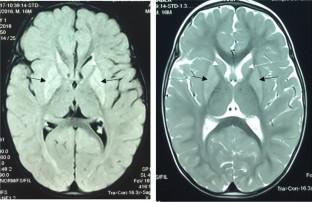
Short-chain enoyl-CoA hydratase deficiency (ECHS1D) is a rare congenital metabolic disorder that follows an autosomal recessive inheritance pattern. It is caused by mutations in the ECHS1 gene, which encodes a mitochondrial enzyme involved in the second step of mitochondrial β-oxidation of fatty acids. The main characteristics of the disease are severe developmental delay, regression, seizures, neurodegeneration, high blood lactate, and a brain MRI pattern consistent with Leigh syndrome. Here, we report three patients belonging to a consanguineous family who presented with mitochondrial encephalomyopathy. Whole-exome sequencing revealed a new homozygous mutation c.619G > A (p.Gly207Ser) at the last nucleotide position in exon 5 of the ECHS1 gene. Experimental analysis showed that normal ECHS1 pre-mRNA splicing occurred in all patients compared to controls. Furthermore, three-dimensional models of wild-type and mutant echs1 proteins revealed changes in catalytic site interactions, conformational changes, and intramolecular interactions, potentially disrupting echs1 protein trimerization and affecting its function. Additionally, the quantification of mtDNA copy number variation in blood leukocytes showed severe mtDNA depletion in all probands.
中文翻译:
短链烯酰辅酶 A 水合酶缺陷和 Mt-DNA 缺失的近亲家族中新型 ECHS1 基因突变的分子和计算机研究:对三聚体组装和催化活性的影响
短链烯酰辅酶A水合酶缺乏症(ECHS1D)是一种罕见的先天性代谢性疾病,遵循常染色体隐性遗传模式。它是由ECHS1基因突变引起的,该基因编码参与线粒体脂肪酸 β-氧化第二步的线粒体酶。该疾病的主要特征是严重的发育迟缓、退化、癫痫发作、神经退行性变、高血乳酸以及与 Leigh 综合征一致的脑部 MRI 模式。在这里,我们报告了三名来自近亲家庭的患有线粒体脑肌病的患者。全外显子组测序揭示了ECHS1基因外显子 5 的最后一个核苷酸位置处存在新的纯合突变 c.619G > A (p.Gly207Ser)。实验分析表明,与对照组相比,所有患者均出现正常的ECHS1 mRNA 前体剪接。此外,野生型和突变型 echs1 蛋白的三维模型揭示了催化位点相互作用、构象变化和分子内相互作用的变化,可能破坏 echs1 蛋白三聚化并影响其功能。此外,血液白细胞中 mtDNA 拷贝数变异的量化显示所有先证者的 mtDNA 严重缺失。


























 京公网安备 11010802027423号
京公网安备 11010802027423号