Molecular Genetics and Genomics ( IF 3.1 ) Pub Date : 2024-02-21 , DOI: 10.1007/s00438-024-02097-7 Leo A. Featherstone , Angela McGaughran
|
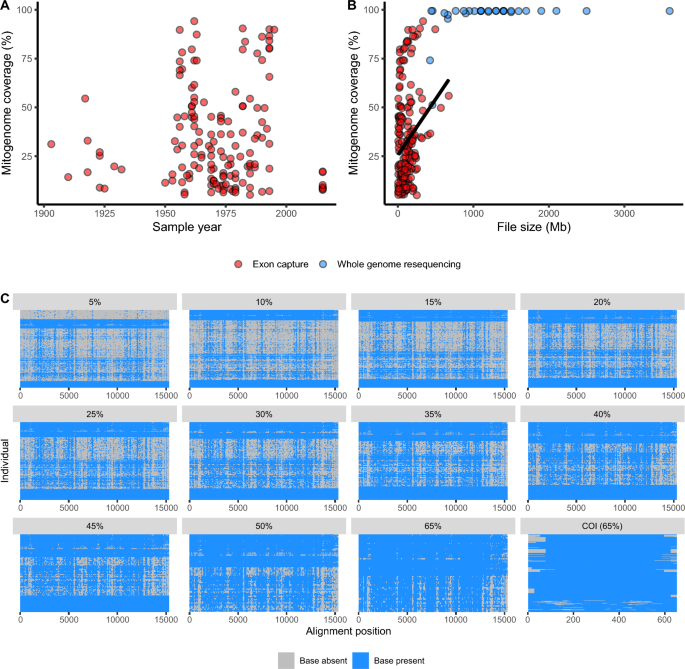
Sequence capture is a genomic technique that selectively enriches target sequences before high throughput next-generation sequencing, to generate specific sequences of interest. Off-target or ‘bycatch’ data are often discarded from capture experiments, but can be leveraged to address evolutionary questions under some circumstances. Here, we investigated the effects of missing data on a variety of evolutionary analyses using bycatch from an exon capture experiment on the global pest moth, Helicoverpa armigera. We added > 200 new samples from across Australia in the form of mitogenomes obtained as bycatch from targeted sequence capture, and combined these into an additional larger dataset to total > 1000 mitochondrial cytochrome c oxidase subunit I (COI) sequences across the species’ global distribution. Using discriminant analysis of principal components and Bayesian coalescent analyses, we showed that mitogenomes assembled from bycatch with up to 75% missing data were able to return evolutionary inferences consistent with higher coverage datasets and the broader literature surrounding H. armigera. For example, low-coverage sequences broadly supported the delineation of two H. armigera subspecies and also provided new insights into the potential for geographic turnover among these subspecies. However, we also identified key effects of dataset coverage and composition on our results. Thus, low-coverage bycatch data can offer valuable information for population genetic and phylodynamic analyses, but caution is required to ensure the reduced information does not introduce confounding factors, such as sampling biases, that drive inference. We encourage more researchers to consider maximizing the potential of the targeted sequence approach by examining evolutionary questions with their off-target bycatch where possible—especially in cases where no previous mitochondrial data exists—but recommend stratifying data at different genome coverage thresholds to separate sampling effects from genuine genomic signals, and to understand their implications for evolutionary research.
中文翻译:
缺失数据对序列捕获兼捕物进化分析的影响及其在农业害虫中的应用
序列捕获是一种基因组技术,可在高通量下一代测序之前选择性地富集目标序列,以生成感兴趣的特定序列。脱靶或“兼捕”数据通常在捕获实验中被丢弃,但在某些情况下可以用来解决进化问题。在这里,我们利用全球害虫棉铃虫外显子捕获实验中的副渔获物,研究了缺失数据对各种进化分析的影响。我们添加了来自澳大利亚各地的超过 200 个新样本,这些样本以从目标序列捕获中捕获的副渔获物形式获得,并将这些样本组合成一个更大的数据集,从而在该物种的全球分布中总计超过 1000 个线粒体细胞色素C氧化酶亚基 I (COI) 序列。通过对主成分的判别分析和贝叶斯合并分析,我们发现,由缺失数据高达 75% 的副渔获物组装的有丝分裂基因组能够返回与更高覆盖率数据集和有关棉铃虫的更广泛文献一致的进化推论。例如,低覆盖率序列广泛支持了两个棉铃虫亚种的划分,也为这些亚种之间地理更替的潜力提供了新的见解。然而,我们还确定了数据集覆盖范围和组成对我们结果的关键影响。因此,低覆盖率兼捕数据可以为种群遗传和系统动力学分析提供有价值的信息,但需要谨慎确保减少的信息不会引入干扰因素,例如导致推论的抽样偏差。我们鼓来自真正的基因组信号,并了解它们对进化研究的影响。


























 京公网安备 11010802027423号
京公网安备 11010802027423号