当前位置:
X-MOL 学术
›
Chemistryopen
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Studying the adsorption of emerging organic contaminants in zeolites with dispersion‐corrected density functional theory calculations: From numbers to recommendations
ChemistryOpen ( IF 2.3 ) Pub Date : 2024-02-22 , DOI: 10.1002/open.202300273 Michael Fischer 1, 2 , Jakob Brauer 1, 2
ChemistryOpen ( IF 2.3 ) Pub Date : 2024-02-22 , DOI: 10.1002/open.202300273 Michael Fischer 1, 2 , Jakob Brauer 1, 2
Affiliation
|
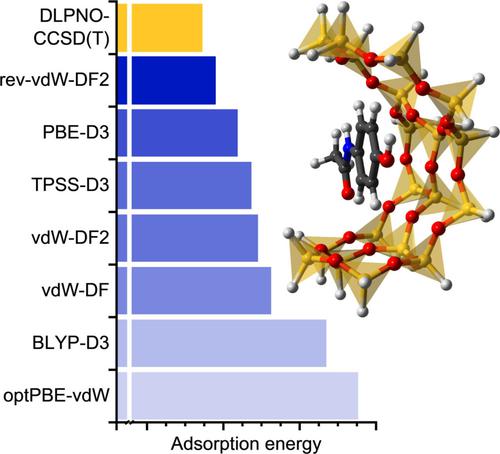
Adsorption energies obtained from dispersion‐corrected density functional theory (DFT) calculations show a considerable dependence on the choice of exchange‐correlation functional and dispersion correction. A number of investigations have employed different approaches to compute adsorption energies of small molecules in zeolites, using reference values from high‐level calculations and/or experiments. Such comparative studies are lacking for larger functional organic molecules such as pharmaceuticals or personal care products, despite their potential relevance for applications, e. g ., in contaminant removal or drug delivery. The present study aims to fill this gap by comparing adsorption energies and, for selected cases, equilibrium structures of emerging organic contaminants adsorbed in MOR‐ and FAU‐type all‐silica zeolites. A total of 13 dispersion‐corrected DFT approaches are compared, including methods using a pairwise dispersion correction as well as non‐local van der Waals density functionals. While absolute values of adsorption energies vary widely, qualitative trends across the set of zeolite‐guest combinations are not strongly dependent on the choice of functional. For selected cluster models, DFT adsorption energies are compared to reference values from coupled cluster (DLPNO‐CCSD(T)) calculations. Although all DFT approaches deliver systematically more negative adsorption energies than the coupled cluster reference, this tendency is least pronounced for the rev‐vdW‐DF2 functional.
中文翻译:

通过色散校正密度泛函理论计算研究沸石中新兴有机污染物的吸附:从数字到建议
从色散校正密度泛函理论(DFT)计算获得的吸附能显示出很大程度上依赖于交换相关函数和色散校正的选择。许多研究采用不同的方法来计算沸石中小分子的吸附能,并使用高级计算和/或实验的参考值。尽管药物或个人护理产品等较大的功能性有机分子具有潜在的应用相关性,但此类比较研究仍然缺乏。e. G .,在污染物去除或药物输送中。本研究旨在通过比较吸附能以及在选定情况下吸附在 MOR 型和 FAU 型全硅沸石中的新兴有机污染物的平衡结构来填补这一空白。总共比较了 13 种色散校正 DFT 方法,包括使用成对色散校正以及非局部范德华密度泛函的方法。虽然吸附能的绝对值差异很大,但沸石-客体组合的定性趋势并不强烈依赖于功能的选择。对于选定的簇模型,将 DFT 吸附能与耦合簇 (DLPNO-CCSD(T)) 计算的参考值进行比较。尽管所有 DFT 方法都比耦合簇参考系统提供更多的负吸附能,但这种趋势对于 rev-vdW-DF2 泛函来说最不明显。
更新日期:2024-02-22
中文翻译:
通过色散校正密度泛函理论计算研究沸石中新兴有机污染物的吸附:从数字到建议
从色散校正密度泛函理论(DFT)计算获得的吸附能显示出很大程度上依赖于交换相关函数和色散校正的选择。许多研究采用不同的方法来计算沸石中小分子的吸附能,并使用高级计算和/或实验的参考值。尽管药物或个人护理产品等较大的功能性有机分子具有潜在的应用相关性,但此类比较研究仍然缺乏。


























 京公网安备 11010802027423号
京公网安备 11010802027423号