当前位置:
X-MOL 学术
›
Bioorgan. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Benzimidazole-oxindole hybrids as multi-kinase inhibitors targeting melanoma
Bioorganic Chemistry ( IF 5.1 ) Pub Date : 2024-02-26 , DOI: 10.1016/j.bioorg.2024.107243 Rasha M. Allam , Ahmed M. El Kerdawy , Ahmed Gouda , Kawkab A. Ahmed , Heba T. Abdel-Mohsen
Bioorganic Chemistry ( IF 5.1 ) Pub Date : 2024-02-26 , DOI: 10.1016/j.bioorg.2024.107243 Rasha M. Allam , Ahmed M. El Kerdawy , Ahmed Gouda , Kawkab A. Ahmed , Heba T. Abdel-Mohsen
|
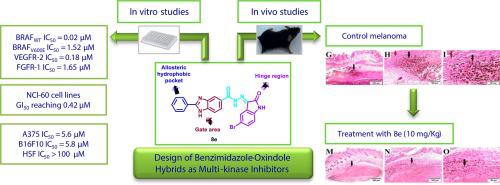
In the current study, a series of benzimidazole-oxindole conjugates were designed and synthesized as type II multi-kinase inhibitors. They exhibited moderate to potent inhibitory activity against BRAF up to 99.61 % at 10 µM. Notably, compounds and demonstrated the most promising activity, with 99.44 to 99.61 % inhibition. Further evaluation revealed that and exhibit moderate to potent inhibitory effects on the kinases BRAF, VEGFR-2, and FGFR-1. Additionally, compounds were screened for their cytotoxicity by the NCI, and several compounds showed significant growth inhibition in diverse cancer cell lines. Compound stood out with a GI range of 1.23 – 3.38 µM on melanoma cell lines. Encouraged by its efficacy, it was further investigated for its antitumor activity and mechanism of action, using sorafenib as a reference standard. The hybrid compound exhibited potent cellular-level suppression of BRAF, VEGFR-2, and FGFR-1 in A375 cell line, surpassing the effects of sorafenib. studies demonstrate that significantly inhibits the growth of B16F10 tumors in mice, leading to increased survival rates and histopathological tumor regression. Furthermore, reduces angiogenesis markers, mRNA expression levels of VEGFR-2 and FGFR-1, and production of growth factors. It also downregulated Notch1 protein expression and decreased TGF-β1 production. Molecular docking simulations suggest that binds as a promising type II kinase inhibitor in the target kinases interacting with the key regions in their kinase domain.
中文翻译:

苯并咪唑-羟吲哚杂合体作为针对黑色素瘤的多激酶抑制剂
在本研究中,设计并合成了一系列苯并咪唑-羟吲哚缀合物作为II型多激酶抑制剂。它们在 10 µM 浓度下对 BRAF 表现出中度至强效的抑制活性,高达 99.61%。值得注意的是,化合物 和 表现出最有希望的活性,抑制率为 99.44% 至 99.61%。进一步的评估表明,对激酶 BRAF、VEGFR-2 和 FGFR-1 表现出中度至强效的抑制作用。此外,NCI 对化合物的细胞毒性进行了筛选,一些化合物在不同的癌细胞系中表现出显着的生长抑制作用。该化合物在黑色素瘤细胞系上的 GI 范围为 1.23 – 3.38 µM。受其功效的鼓舞,以索拉非尼为参考标准,进一步研究其抗肿瘤活性和作用机制。该混合化合物在 A375 细胞系中表现出对 BRAF、VEGFR-2 和 FGFR-1 的有效细胞水平抑制,超过了索拉非尼的效果。研究表明,它能显着抑制小鼠 B16F10 肿瘤的生长,从而提高存活率和组织病理学肿瘤消退。此外,还可降低血管生成标记物、VEGFR-2 和 FGFR-1 的 mRNA 表达水平以及生长因子的产生。它还下调了 Notch1 蛋白的表达并减少了 TGF-β1 的产生。分子对接模拟表明,作为一种有前景的 II 型激酶抑制剂,可与靶激酶结合,与其激酶结构域中的关键区域相互作用。
更新日期:2024-02-26
中文翻译:
苯并咪唑-羟吲哚杂合体作为针对黑色素瘤的多激酶抑制剂
在本研究中,设计并合成了一系列苯并咪唑-羟吲哚缀合物作为II型多激酶抑制剂。它们在 10 µM 浓度下对 BRAF 表现出中度至强效的抑制活性,高达 99.61%。值得注意的是,化合物 和 表现出最有希望的活性,抑制率为 99.44% 至 99.61%。进一步的评估表明,对激酶 BRAF、VEGFR-2 和 FGFR-1 表现出中度至强效的抑制作用。此外,NCI 对化合物的细胞毒性进行了筛选,一些化合物在不同的癌细胞系中表现出显着的生长抑制作用。该化合物在黑色素瘤细胞系上的 GI 范围为 1.23 – 3.38 µM。受其功效的鼓舞,以索拉非尼为参考标准,进一步研究其抗肿瘤活性和作用机制。该混合化合物在 A375 细胞系中表现出对 BRAF、VEGFR-2 和 FGFR-1 的有效细胞水平抑制,超过了索拉非尼的效果。研究表明,它能显着抑制小鼠 B16F10 肿瘤的生长,从而提高存活率和组织病理学肿瘤消退。此外,还可降低血管生成标记物、VEGFR-2 和 FGFR-1 的 mRNA 表达水平以及生长因子的产生。它还下调了 Notch1 蛋白的表达并减少了 TGF-β1 的产生。分子对接模拟表明,作为一种有前景的 II 型激酶抑制剂,可与靶激酶结合,与其激酶结构域中的关键区域相互作用。


























 京公网安备 11010802027423号
京公网安备 11010802027423号