当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
A Charge–Charge Flux–Dipole Flux Analysis of Simple Molecular Systems with Halogen Bonds
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2024-03-08 , DOI: 10.1021/acs.jpca.3c08229 Paulo Eduardo Cavalcanti Martins Filho 1 , Roberto Luiz Andrade Haiduke 1
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2024-03-08 , DOI: 10.1021/acs.jpca.3c08229 Paulo Eduardo Cavalcanti Martins Filho 1 , Roberto Luiz Andrade Haiduke 1
Affiliation
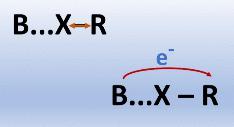
|
The presence of halogen bonds (R–X···B; R = substituent group, X = halogen, and B = Lewis base) provides quite amazing molecular systems for electronic structure investigations, presenting unique characteristics of fundamental relevance to supramolecular chemistry among other areas. Here, we use a double-hybrid approach from Density Functional Theory and triple-ζ basis sets augmented with diffuse functions (B2PLYP/def2-TZVPD) to deal with a large group of simple molecular systems containing halogen bonds (XBs), focusing on geometrical structures, binding energies, harmonic vibrational frequencies, and fundamental infrared intensities. Next, the electron densities and their variations on vibrations are carefully studied with the Quantum Theory of Atoms in Molecules (QTAIM) formalism and the charge–charge flux–dipole flux (CCFDF) model. We notice that the R–X stretching mode usually shows vibrational frequency decrements and infrared intensifications during the XB formation. Such features were also observed in hydrogen bonds, although the explanation for the band strengthening is different. Surprisingly, the most important contribution to these intensity increments due to complexation is now the interaction term between the charge flux and dipole flux (CF × DF). Thus, the use of atomic dipoles is mandatory to fully understand this phenomenon. In fact, the huge charge flux contributions to changes in dipole moment derivatives of R–X stretchings on halogen bonding are no longer accompanied by opposite variations of similar magnitudes in polarizations described by atomic dipole fluxes, which provided nearly unaltered values during the XB formation. Thus, the electronic charge flux direction change that takes place in complexes (from B to R) now reinforces dipole moment derivative terms from such atomic polarizations (mainly from the X atom). This intermolecular charge flux seems to be responsible for the unusual features noticed in the R–X stretching mode with the CCDDF/QTAIM model.
中文翻译:

具有卤素键的简单分子系统的电荷-电荷通量-偶极子通量分析
卤素键(R–X···B;R = 取代基,X = 卤素,B = 路易斯碱)的存在为电子结构研究提供了非常令人惊奇的分子系统,呈现出与超分子化学等基本相关的独特特征地区。在这里,我们使用密度泛函理论的双混合方法和用扩散函数增强的三重 z 基组 (B2PLYP/def2-TZVPD) 来处理一大组包含卤素键 (XB) 的简单分子系统,重点关注几何结构、结合能、简谐振动频率和基本红外强度。接下来,利用分子中原子量子理论 (QTAIM) 形式和电荷-电荷通量-偶极子通量 (CCFDF) 模型仔细研究电子密度及其对振动的变化。我们注意到,R-X 拉伸模式通常在 XB 形成过程中表现出振动频率减小和红外增强。尽管对能带强化的解释不同,但在氢键中也观察到了这种特征。令人惊讶的是,由于络合作用对这些强度增量的最重要贡献现在是电荷通量和偶极通量(CF × DF)之间的相互作用项。因此,为了充分理解这种现象,必须使用原子偶极子。事实上,对卤素键上 R-X 拉伸的偶极矩导数变化的巨大电荷通量贡献不再伴随着原子偶极通量描述的极化中类似幅度的相反变化,这在 XB 形成过程中提供了几乎不变的值。因此,复合物中发生的电子电荷通量方向变化(从 B 到 R)现在强化了来自此类原子极化(主要来自 X 原子)的偶极矩导数项。这种分子间电荷通量似乎是 CCDDF/QTAIM 模型的 R-X 拉伸模式中注意到的异常特征的原因。
更新日期:2024-03-08
中文翻译:
具有卤素键的简单分子系统的电荷-电荷通量-偶极子通量分析
卤素键(R–X···B;R = 取代基,X = 卤素,B = 路易斯碱)的存在为电子结构研究提供了非常令人惊奇的分子系统,呈现出与超分子化学等基本相关的独特特征地区。在这里,我们使用密度泛函理论的双混合方法和用扩散函数增强的三重 z 基组 (B2PLYP/def2-TZVPD) 来处理一大组包含卤素键 (XB) 的简单分子系统,重点关注几何结构、结合能、简谐振动频率和基本红外强度。接下来,利用分子中原子量子理论 (QTAIM) 形式和电荷-电荷通量-偶极子通量 (CCFDF) 模型仔细研究电子密度及其对振动的变化。我们注意到,R-X 拉伸模式通常在 XB 形成过程中表现出振动频率减小和红外增强。尽管对能带强化的解释不同,但在氢键中也观察到了这种特征。令人惊讶的是,由于络合作用对这些强度增量的最重要贡献现在是电荷通量和偶极通量(CF × DF)之间的相互作用项。因此,为了充分理解这种现象,必须使用原子偶极子。事实上,对卤素键上 R-X 拉伸的偶极矩导数变化的巨大电荷通量贡献不再伴随着原子偶极通量描述的极化中类似幅度的相反变化,这在 XB 形成过程中提供了几乎不变的值。因此,复合物中发生的电子电荷通量方向变化(从 B 到 R)现在强化了来自此类原子极化(主要来自 X 原子)的偶极矩导数项。这种分子间电荷通量似乎是 CCDDF/QTAIM 模型的 R-X 拉伸模式中注意到的异常特征的原因。


























 京公网安备 11010802027423号
京公网安备 11010802027423号