Journal of Computer-Aided Molecular Design ( IF 3.5 ) Pub Date : 2024-03-12 , DOI: 10.1007/s10822-024-00557-1 Azam Nesabi , Jas Kalayan , Sara Al-Rawashdeh , Mohammad A. Ghattas , Richard A. Bryce
|
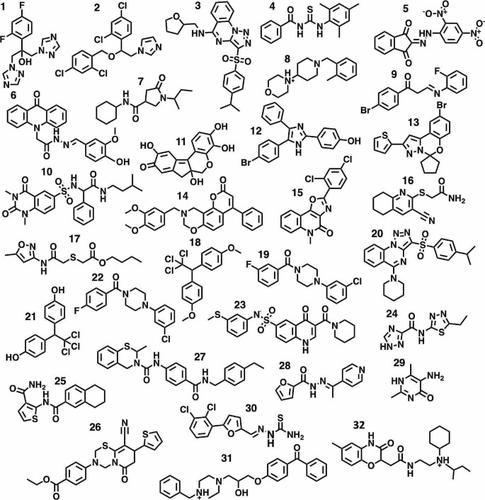
Small colloidally aggregating molecules (SCAMs) can be problematic for biological assays in drug discovery campaigns. However, the self-associating properties of SCAMs have potential applications in drug delivery and analytical biochemistry. Consequently, the ability to predict the aggregation propensity of a small organic molecule is of considerable interest. Chemoinformatics-based filters such as ChemAGG and Aggregator Advisor offer rapid assessment but are limited by the assay quality and structural diversity of their training set data. Complementary to these tools, we explore here the ability of molecular dynamics (MD) simulations as a physics-based method capable of predicting the aggregation propensity of diverse chemical structures. For a set of 32 molecules, using simulations of 100 ns in explicit solvent, we find a success rate of 97% (one molecule misclassified) as opposed to 75% by Aggregator Advisor and 72% by ChemAGG. These short timescale MD simulations are representative of longer microsecond trajectories and yield an informative spectrum of aggregation propensities across the set of solutes, capturing the dynamic behaviour of weakly aggregating compounds. Implicit solvent simulations using the generalized Born model were less successful in predicting aggregation propensity. MD simulations were also performed to explore structure-aggregation relationships for selected molecules, identifying chemical modifications that reversed the predicted behaviour of a given aggregator/non-aggregator compound. While lower throughput than rapid cheminformatics-based SCAM filters, MD-based prediction of aggregation has potential to be deployed on the scale of focused subsets of moderate size, and, depending on the target application, provide guidance on removing or optimizing a compound’s aggregation propensity.
Graphical Abstract
中文翻译:
分子动力学模拟作为调节小分子聚集的指南
小胶体聚集分子 (SCAM) 可能会给药物发现活动中的生物测定带来问题。然而,SCAM 的自缔合特性在药物输送和分析生物化学方面具有潜在的应用。因此,预测有机小分子聚集倾向的能力引起了人们的极大兴趣。基于化学信息学的过滤器(例如 ChemAGG 和 Aggregator Advisor)可提供快速评估,但受到其训练集数据的检测质量和结构多样性的限制。作为这些工具的补充,我们在此探索分子动力学(MD)模拟作为一种基于物理的方法的能力,能够预测不同化学结构的聚集倾向。对于一组 32 个分子,在显式溶剂中使用 100 ns 的模拟,我们发现成功率为 97%(一个分子被错误分类),而 Aggregator Advisor 为 75%,ChemAGG 为 72%。这些短时间尺度的 MD 模拟代表了较长的微秒轨迹,并产生了一组溶质的聚集倾向的信息谱,捕获了弱聚集化合物的动态行为。使用广义 Born 模型的隐式溶剂模拟在预测聚集倾向方面不太成功。还进行MD模拟来探索所选分子的结构-聚集关系,识别可逆转给定聚集剂/非聚集剂化合物的预测行为的化学修饰。虽然吞吐量低于基于化学信息学的快速 SCAM 过滤器,但基于 MD 的聚集预测有可能在中等大小的集中子集的规模上部署,并且根据目标应用,为消除或优化化合物的聚集倾向提供指导。


























 京公网安备 11010802027423号
京公网安备 11010802027423号