当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Performance of EOM-CCSD(T)(a)*-Based Quartic Force Fields in Computing Fundamental, Anharmonic Vibrational Frequencies of Molecular Electronically Excited States with Application to the Ã1A″ State of :CCH2 (Vinylidene)
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2024-03-11 , DOI: 10.1021/acs.jpca.3c08168 Alexandria G. Watrous 1 , Megan C. Davis 1 , Ryan C. Fortenberry 1
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2024-03-11 , DOI: 10.1021/acs.jpca.3c08168 Alexandria G. Watrous 1 , Megan C. Davis 1 , Ryan C. Fortenberry 1
Affiliation
|
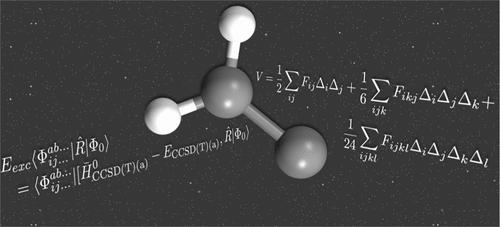
Highly accurate anharmonic vibrational frequencies of electronically excited states are not as easily computed as their ground electronic state counterparts, but recently developed approximate triple excited state methods may be changing that. One emerging excited state method is equation of motion coupled cluster theory at the singles and doubles level with perturbative triples computed via the (a)* formalism, EOMEE-CCSD(T)(a)*. One of the most employed means for the ready computation of vibrational anharmonic frequencies for ground electronic states is second-order vibrational perturbation theory (VPT2), a theory based on quartic force fields (QFFs),fourth-order Taylor series expansions of the potential portion of the internuclear Watson Hamiltonian. The combination of these two is herein benchmarked for its performance for use as a means of computing rovibrational spectra of electronically excited states. Specifically, the EOMEE-CCSD(T)(a)* approach employing a complete basis set extrapolation along with core electron inclusion and relativity (the so-called “CcCR” approach) defining the QFF produces anharmonic fundamental vibrational frequencies within 2.83%, on the average, of reported gas-phase experimentally assigned values for the test set including the ˜A1A″A~1A′′ states of HCF, HCCl, HSiF, HNO, and HPO. However, some states have exceptional accuracy in the fundamentals, most notably for ν2 of ˜A1A″A~1A′′ HCCl in which the CcCR QFF value is within 1.8 cm–1 at 927.9 cm–1 (or 0.2%) of the experiment. Additionally, this approach produces rotational constants to, on the absolute average, within 0.41% of available experimental data, showcasing notable accuracy in the computation of rovibronic spectral data. Furthermore, utilizing a hybrid approach composed of harmonic CcCR force constants along with a set of simple EOMEE-CCSD(T)(a)*/aug-cc-pVQZ QFF cubic and quartic force constants is faster than using pure CcCR and better represents those modes that suffer from numerical instability in the anharmonic portion of the QFF, implying that this so-called “CcCR + QZ” QFF approach may be the best for future applications. Finally, complete, rovibrational spectral data are provided for ˜A1A2A~1A2 :CCH2, a molecule of potential astrochemical interest, in order to aid in its potential future experimental rovibronic characterization.
中文翻译:

基于 EOM-CCSD(T)(a)* 的四次力场在计算分子电子激发态的基本非简振动频率中的性能,并应用于 :CCH2(亚乙烯)的 à1A" 态
电子激发态的高精度非谐振动频率并不像其基态电子态对应物那样容易计算,但最近开发的近似三激发态方法可能正在改变这一点。一种新兴的激发态方法是单打和双打水平上的运动方程耦合簇理论,通过 (a)* 形式 EOMEE-CCSD(T)(a)* 计算微扰三元组。用于快速计算基态电子态振动非谐频率的最常用方法之一是二阶振动扰动理论(VPT2),这是一种基于四次力场(QFF)、势部分的四阶泰勒级数展开的理论核间沃森哈密顿量。本文对这两者的组合作为计算电子激发态的振动光谱的手段的性能进行了基准测试。具体来说,EOMEE-CCSD(T)(a)* 方法采用完整的基组外推以及核心电子包含和相对论(所谓的“CcCR”方法)来定义 QFF,产生 2.83% 以内的非谐基频振动频率,测试集报告的气相实验指定值的平均值,包括~A 1A”A~1A′′ HCF、HCl、HSiF、HNO 和 HPO 的状态。然而,一些状态在基本原理上具有异常的准确性,尤其是对于ν 2~A 1A”A~1A′′ HCl,其中 CcCR QFF 值在实验的927.9 cm –1处(或 0.2%)在 1.8 cm –1以内。此外,这种方法产生的旋转常数的绝对平均值在可用实验数据的 0.41% 以内,显示了 rovibronic 光谱数据计算的显着准确性。此外,利用由谐波 CcCR 力常数以及一组简单的 EOMEE-CCSD(T)(a)*/aug-cc-pVQZ QFF 三次和四次力常数组成的混合方法比使用纯 CcCR 更快,并且更好地表示这些力常数QFF 的非谐波部分存在数值不稳定的模式,这意味着这种所谓的“CcCR + QZ”QFF 方法可能是未来应用的最佳选择。最后,提供了完整的旋转光谱数据〜A 1A2A~1A2 :CCH 2,一种具有潜在天体化学意义的分子,以帮助其未来潜在的实验性振动电子表征。
更新日期:2024-03-11
中文翻译:
基于 EOM-CCSD(T)(a)* 的四次力场在计算分子电子激发态的基本非简振动频率中的性能,并应用于 :CCH2(亚乙烯)的 à1A" 态
电子激发态的高精度非谐振动频率并不像其基态电子态对应物那样容易计算,但最近开发的近似三激发态方法可能正在改变这一点。一种新兴的激发态方法是单打和双打水平上的运动方程耦合簇理论,通过 (a)* 形式 EOMEE-CCSD(T)(a)* 计算微扰三元组。用于快速计算基态电子态振动非谐频率的最常用方法之一是二阶振动扰动理论(VPT2),这是一种基于四次力场(QFF)、势部分的四阶泰勒级数展开的理论核间沃森哈密顿量。本文对这两者的组合作为计算电子激发态的振动光谱的手段的性能进行了基准测试。具体来说,EOMEE-CCSD(T)(a)* 方法采用完整的基组外推以及核心电子包含和相对论(所谓的“CcCR”方法)来定义 QFF,产生 2.83% 以内的非谐基频振动频率,测试集报告的气相实验指定值的平均值,包括~A 1A”


























 京公网安备 11010802027423号
京公网安备 11010802027423号