当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Transferability of Machine Learning Models for Predicting Raman Spectra
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2024-03-13 , DOI: 10.1021/acs.jpca.3c07109 Mandi Fang 1, 2 , Shi Tang 2 , Zheyong Fan 3 , Yao Shi 1 , Nan Xu 1, 2 , Yi He 1, 2, 4
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2024-03-13 , DOI: 10.1021/acs.jpca.3c07109 Mandi Fang 1, 2 , Shi Tang 2 , Zheyong Fan 3 , Yao Shi 1 , Nan Xu 1, 2 , Yi He 1, 2, 4
Affiliation
|
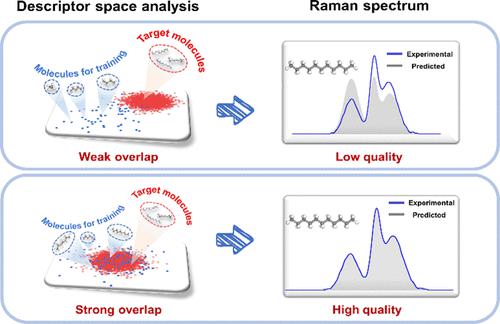
Theoretical prediction of vibrational Raman spectra enables a detailed interpretation of experimental spectra, and the advent of machine learning techniques makes it possible to predict Raman spectra while achieving a good balance between efficiency and accuracy. However, the transferability of machine learning models across different molecules remains poorly understood. This work proposed a new strategy whereby machine learning-based polarizability models were trained on similar but smaller alkane molecules to predict spectra of larger alkanes, avoiding extensive first-principles calculations on certain systems. Results showed that the developed polarizability model for alkanes with a maximum of nine carbon atoms can exhibit high accuracy in the predictions of polarizabilities and Raman spectra for the n-undecane molecule (11 carbon atoms), validating its reasonable extrapolation capability. Additionally, a descriptor space analysis method was further introduced to evaluate the transferability, demonstrating potentials for accurate and efficient Raman predictions of large molecules using limited training data labeled for smaller molecules.
中文翻译:

用于预测拉曼光谱的机器学习模型的可迁移性
振动拉曼光谱的理论预测可以对实验光谱进行详细解释,而机器学习技术的出现使得预测拉曼光谱成为可能,同时在效率和准确性之间取得良好的平衡。然而,机器学习模型在不同分子之间的可迁移性仍然知之甚少。这项工作提出了一种新策略,即在相似但较小的烷烃分子上训练基于机器学习的极化率模型,以预测较大烷烃的光谱,从而避免在某些系统上进行大量的第一原理计算。结果表明,所建立的最多9个碳原子的烷烃极化率模型对正十一烷分子(11个碳原子)的极化率和拉曼光谱的预测具有较高的准确性,验证了其合理的外推能力。此外,还引入了描述符空间分析方法来评估可转移性,证明了使用标记为小分子的有限训练数据准确有效地预测大分子拉曼的潜力。
更新日期:2024-03-13
中文翻译:
用于预测拉曼光谱的机器学习模型的可迁移性
振动拉曼光谱的理论预测可以对实验光谱进行详细解释,而机器学习技术的出现使得预测拉曼光谱成为可能,同时在效率和准确性之间取得良好的平衡。然而,机器学习模型在不同分子之间的可迁移性仍然知之甚少。这项工作提出了一种新策略,即在相似但较小的烷烃分子上训练基于机器学习的极化率模型,以预测较大烷烃的光谱,从而避免在某些系统上进行大量的第一原理计算。结果表明,所建立的最多9个碳原子的烷烃极化率模型对正十一烷分子(11个碳原子)的极化率和拉曼光谱的预测具有较高的准确性,验证了其合理的外推能力。此外,还引入了描述符空间分析方法来评估可转移性,证明了使用标记为小分子的有限训练数据准确有效地预测大分子拉曼的潜力。


























 京公网安备 11010802027423号
京公网安备 11010802027423号