当前位置:
X-MOL 学术
›
Acta Pharm. Sin. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
ASF1A-dependent P300-mediated histone H3 lysine 18 lactylation promotes atherosclerosis by regulating EndMT
Acta Pharmaceutica Sinica B ( IF 14.5 ) Pub Date : 2024-03-12 , DOI: 10.1016/j.apsb.2024.03.008 Mengdie Dong , Yunjia Zhang , Minghong Chen , Yongkang Tan , Jiao Min , Xian He , Fuhao Liu , Jiaming Gu , Hong Jiang , Longbin Zheng , Jiajing Chen , Quanwen Yin , Xuesong Li , Xiang Chen , Yongfeng Shao , Yong Ji , Hongshan Chen
Acta Pharmaceutica Sinica B ( IF 14.5 ) Pub Date : 2024-03-12 , DOI: 10.1016/j.apsb.2024.03.008 Mengdie Dong , Yunjia Zhang , Minghong Chen , Yongkang Tan , Jiao Min , Xian He , Fuhao Liu , Jiaming Gu , Hong Jiang , Longbin Zheng , Jiajing Chen , Quanwen Yin , Xuesong Li , Xiang Chen , Yongfeng Shao , Yong Ji , Hongshan Chen
|
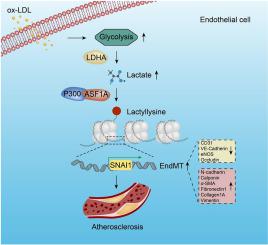
Endothelial-to-mesenchymal transition (EndMT) is a key driver of atherosclerosis. Aerobic glycolysis is increased in the endothelium of atheroprone areas, accompanied by elevated lactate levels. Histone lactylation, mediated by lactate, can regulate gene expression and participate in disease regulation. However, whether histone lactylation is involved in atherosclerosis remains unknown. Here, we report that lipid peroxidation could lead to EndMT-induced atherosclerosis by increasing lactate-dependent histone H3 lysine 18 lactylation (H3K18la) and , as well as in atherosclerotic patients’ arteries. Mechanistically, the histone chaperone ASF1A was first identified as a cofactor of P300, which precisely regulated the enrichment of H3K18la at the promoter of , thereby activating transcription and promoting EndMT. We found that deletion of ASF1A inhibited EndMT and improved endothelial dysfunction. Functional analysis based on mice in the atherosclerosis model confirmed the involvement of H3K18la in atherosclerosis and found that endothelium-specific ASF1A deficiency inhibited EndMT and alleviated atherosclerosis development. Inhibition of glycolysis by pharmacologic inhibition and advanced PROTAC attenuated H3K18la, transcription, and EndMT-induced atherosclerosis. This study illustrates precise crosstalk between metabolism and epigenetics H3K18la by the P300/ASF1A molecular complex during EndMT-induced atherogenesis, which provides emerging therapies for atherosclerosis.
中文翻译:

ASF1A依赖性P300介导的组蛋白H3赖氨酸18乳酰化通过调节EndMT促进动脉粥样硬化
内皮-间质转化(EndMT)是动脉粥样硬化的关键驱动因素。动脉粥样硬化易发区域内皮细胞的有氧糖酵解增加,并伴有乳酸水平升高。由乳酸介导的组蛋白乳酰化可以调节基因表达并参与疾病调节。然而,组蛋白乳酰化是否与动脉粥样硬化有关仍不清楚。在此,我们报道脂质过氧化可能通过增加乳酸依赖性组蛋白 H3 赖氨酸 18 乳酰化 (H3K18la) 和动脉粥样硬化患者的动脉,导致 EndMT 诱导的动脉粥样硬化。从机制上来说,组蛋白伴侣ASF1A首先被鉴定为P300的辅助因子,它精确调节H3K18la在启动子处的富集,从而激活转录并促进EndMT。我们发现 ASF1A 的缺失会抑制 EndMT 并改善内皮功能障碍。基于动脉粥样硬化模型小鼠的功能分析证实了 H3K18la 参与动脉粥样硬化,并发现内皮特异性 ASF1A 缺陷可抑制 EndMT 并减轻动脉粥样硬化的发展。通过药理学抑制和先进的 PROTAC 抑制糖酵解可减轻 H3K18la、转录和 EndMT 诱导的动脉粥样硬化。这项研究阐明了 EndMT 诱导的动脉粥样硬化形成过程中 P300/ASF1A 分子复合物代谢与表观遗传学 H3K18la 之间的精确串扰,这为动脉粥样硬化提供了新兴疗法。
更新日期:2024-03-12
中文翻译:
ASF1A依赖性P300介导的组蛋白H3赖氨酸18乳酰化通过调节EndMT促进动脉粥样硬化
内皮-间质转化(EndMT)是动脉粥样硬化的关键驱动因素。动脉粥样硬化易发区域内皮细胞的有氧糖酵解增加,并伴有乳酸水平升高。由乳酸介导的组蛋白乳酰化可以调节基因表达并参与疾病调节。然而,组蛋白乳酰化是否与动脉粥样硬化有关仍不清楚。在此,我们报道脂质过氧化可能通过增加乳酸依赖性组蛋白 H3 赖氨酸 18 乳酰化 (H3K18la) 和动脉粥样硬化患者的动脉,导致 EndMT 诱导的动脉粥样硬化。从机制上来说,组蛋白伴侣ASF1A首先被鉴定为P300的辅助因子,它精确调节H3K18la在启动子处的富集,从而激活转录并促进EndMT。我们发现 ASF1A 的缺失会抑制 EndMT 并改善内皮功能障碍。基于动脉粥样硬化模型小鼠的功能分析证实了 H3K18la 参与动脉粥样硬化,并发现内皮特异性 ASF1A 缺陷可抑制 EndMT 并减轻动脉粥样硬化的发展。通过药理学抑制和先进的 PROTAC 抑制糖酵解可减轻 H3K18la、转录和 EndMT 诱导的动脉粥样硬化。这项研究阐明了 EndMT 诱导的动脉粥样硬化形成过程中 P300/ASF1A 分子复合物代谢与表观遗传学 H3K18la 之间的精确串扰,这为动脉粥样硬化提供了新兴疗法。


























 京公网安备 11010802027423号
京公网安备 11010802027423号