当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Molecular Geometry Impact on Deep Learning Predictions of Inverted Singlet–Triplet Gaps
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2024-03-14 , DOI: 10.1021/acs.jpca.4c00172 Leonardo Barneschi 1 , Leonardo Rotondi 1 , Daniele Padula 1
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2024-03-14 , DOI: 10.1021/acs.jpca.4c00172 Leonardo Barneschi 1 , Leonardo Rotondi 1 , Daniele Padula 1
Affiliation
|
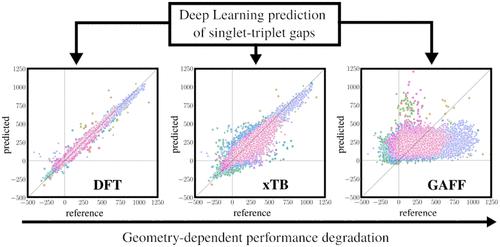
We present a deep learning model able to predict excited singlet–triplet gaps with a mean absolute error (MAE) of ≈20 meV to obtain potential inverted singlet–triplet (IST) candidates. We exploit cutting-edge spherical message passing graph neural networks designed specifically for generating 3D graph representations in molecular learning. In a nutshell, the model takes as input a list of unsaturated heavy atom Cartesian coordinates and atomic numbers, producing singlet–triplet gaps as output. We exploited available large data collections to train the model on ≈40,000 heterogeneous density functional theory (DFT) geometries with available ADC(2)/cc-pVDZ singlet–triplet gaps. We ascertain the predictive power of the model from a quantitative perspective obtaining predictions on a test set of ≈14,000 molecules, whose geometries have been generated at DFT level (the same employed for the geometries in the training set), at GFN2-xTB level, and through Molecular Mechanics. We notice performance degradation upon switching to lower-quality geometries, with GFN2-xTB ones maintaining satisfactory results (MAE ≈ 50 meV on GFN2-xTB geometries, MAE ≈ 180 meV on generalized AMBER force field geometries), hinting at caution when dealing with specific chemical classes. Finally, we verify the performance of the model from the qualitative point of view, obtaining predictions on a different data set of ≈15,000 molecules already used to identify new IST molecules. We obtained predictions using both DFT and experimental X-ray geometries, with results on IST candidates similar to those provided by quantum chemical methods, with clear hints for the path toward improved performance.
中文翻译:

分子几何对倒置单重态-三重态间隙深度学习预测的影响
我们提出了一种深度学习模型,能够预测平均绝对误差 (MAE) 约为 20 meV 的激发单重态 - 三重态间隙,以获得潜在的倒置单重态 - 三重态 (IST) 候选者。我们利用专门为在分子学习中生成 3D 图形表示而设计的尖端球形消息传递图形神经网络。简而言之,该模型将不饱和重原子笛卡尔坐标和原子序数列表作为输入,产生单重态-三重态间隙作为输出。我们利用可用的大量数据集,在 大约 40,000 个异质密度泛函理论 (DFT) 几何结构上训练模型,并具有可用的 ADC(2)/cc-pVDZ 单线态-三线态间隙。我们从定量角度确定模型的预测能力,在 GFN2-xTB 水平上对约 14,000 个分子的测试集进行预测,这些分子的几何形状是在 DFT 级别生成的(与训练集中的几何形状相同),并通过分子力学。我们注意到切换到较低质量的几何形状后性能会下降,而 GFN2-xTB 几何形状保持令人满意的结果(GFN2-xTB 几何形状上的 MAE ≈ 50 meV,广义 AMBER 力场几何形状上的 MAE ≈ 180 meV),这暗示在处理特定的几何形状时要小心化学类。最后,我们从定性的角度验证了模型的性能,获得了对已用于识别新 IST 分子的约 15,000 个分子的不同数据集的预测。我们使用 DFT 和实验 X 射线几何结构获得了预测,IST 候选结果与量子化学方法提供的结果相似,并为提高性能的路径提供了明确的提示。
更新日期:2024-03-14
中文翻译:
分子几何对倒置单重态-三重态间隙深度学习预测的影响
我们提出了一种深度学习模型,能够预测平均绝对误差 (MAE) 约为 20 meV 的激发单重态 - 三重态间隙,以获得潜在的倒置单重态 - 三重态 (IST) 候选者。我们利用专门为在分子学习中生成 3D 图形表示而设计的尖端球形消息传递图形神经网络。简而言之,该模型将不饱和重原子笛卡尔坐标和原子序数列表作为输入,产生单重态-三重态间隙作为输出。我们利用可用的大量数据集,在 大约 40,000 个异质密度泛函理论 (DFT) 几何结构上训练模型,并具有可用的 ADC(2)/cc-pVDZ 单线态-三线态间隙。我们从定量角度确定模型的预测能力,在 GFN2-xTB 水平上对约 14,000 个分子的测试集进行预测,这些分子的几何形状是在 DFT 级别生成的(与训练集中的几何形状相同),并通过分子力学。我们注意到切换到较低质量的几何形状后性能会下降,而 GFN2-xTB 几何形状保持令人满意的结果(GFN2-xTB 几何形状上的 MAE ≈ 50 meV,广义 AMBER 力场几何形状上的 MAE ≈ 180 meV),这暗示在处理特定的几何形状时要小心化学类。最后,我们从定性的角度验证了模型的性能,获得了对已用于识别新 IST 分子的约 15,000 个分子的不同数据集的预测。我们使用 DFT 和实验 X 射线几何结构获得了预测,IST 候选结果与量子化学方法提供的结果相似,并为提高性能的路径提供了明确的提示。


























 京公网安备 11010802027423号
京公网安备 11010802027423号