当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Efficient Method for Numerical Calculations of Molecular Vibrational Frequencies by Exploiting Sparseness of Hessian Matrix
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2024-03-14 , DOI: 10.1021/acs.jpca.3c07645 Xingyu Yang 1, 2 , Haitao Ma 1 , Qing Lu 1 , Wensheng Bian 1, 2
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2024-03-14 , DOI: 10.1021/acs.jpca.3c07645 Xingyu Yang 1, 2 , Haitao Ma 1 , Qing Lu 1 , Wensheng Bian 1, 2
Affiliation
|
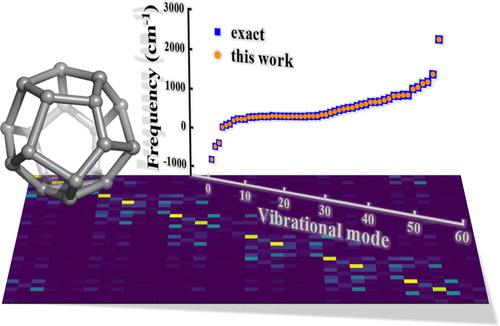
Molecular vibrational frequency analysis plays an important role in theoretical and computational chemistry. However, in many cases, the analytical frequencies are unavailable, whereas frequency calculations using conventional numerical methods are very expensive. In this work, we propose an efficient method to numerically calculate the frequencies. Our main strategies are to exploit the sparseness of the Hessian matrix and to construct the N-fold two-variable potential energy surfaces to fit the parabola parameters, which are later used for the construction of Hessian matrices. A set of benchmark calculations is performed for typical molecules of different sizes and complexities using the proposed method. The obtained frequencies are compared to those calculated with the analytical methods and conventional numerical methods. It is shown that the results yielded with the new method are in very good agreement with corresponding accurate values (with a maximum error of ∼20 cm–1), while the required computation resource is largely reduced compared to that required by conventional numerical methods. For medium-sized molecules, the calculational scaling is lowered to O(N1.6) (this work) from that of O(N2) (conventional numerical methods). For even larger molecules, more computational savings can be achieved, and the scaling is estimated to be quasilinear with respect to the molecular size.
中文翻译:

利用Hessian矩阵稀疏性数值计算分子振动频率的有效方法
分子振动频率分析在理论和计算化学中发挥着重要作用。然而,在许多情况下,无法获得解析频率,而使用传统数值方法计算频率非常昂贵。在这项工作中,我们提出了一种有效的方法来数值计算频率。我们的主要策略是利用Hessian矩阵的稀疏性,构造N重二变量势能面来拟合抛物线参数,随后将其用于Hessian矩阵的构造。使用所提出的方法对不同尺寸和复杂性的典型分子进行一组基准计算。将获得的频率与用解析方法和传统数值方法计算的频率进行比较。结果表明,新方法得到的结果与相应的精确值非常吻合(最大误差为∼20 cm –1),同时与传统数值方法相比,所需的计算资源大大减少。对于中等大小的分子,计算比例从 O(N 2 )(传统数值方法)降低到 O(N 1.6 )(本工作)。对于更大的分子,可以节省更多的计算量,并且缩放比例估计与分子大小呈拟线性关系。
更新日期:2024-03-14
中文翻译:
利用Hessian矩阵稀疏性数值计算分子振动频率的有效方法
分子振动频率分析在理论和计算化学中发挥着重要作用。然而,在许多情况下,无法获得解析频率,而使用传统数值方法计算频率非常昂贵。在这项工作中,我们提出了一种有效的方法来数值计算频率。我们的主要策略是利用Hessian矩阵的稀疏性,构造N重二变量势能面来拟合抛物线参数,随后将其用于Hessian矩阵的构造。使用所提出的方法对不同尺寸和复杂性的典型分子进行一组基准计算。将获得的频率与用解析方法和传统数值方法计算的频率进行比较。结果表明,新方法得到的结果与相应的精确值非常吻合(最大误差为∼20 cm –1),同时与传统数值方法相比,所需的计算资源大大减少。对于中等大小的分子,计算比例从 O(N 2 )(传统数值方法)降低到 O(N 1.6 )(本工作)。对于更大的分子,可以节省更多的计算量,并且缩放比例估计与分子大小呈拟线性关系。


























 京公网安备 11010802027423号
京公网安备 11010802027423号