当前位置:
X-MOL 学术
›
Comput. Struct. Biotechnol. J.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Benchmarking feature selection and feature extraction methods to improve the performances of machine-learning algorithms for patient classification using metabolomics biomedical data
Computational and Structural Biotechnology Journal ( IF 6 ) Pub Date : 2024-03-19 , DOI: 10.1016/j.csbj.2024.03.016 Justine Labory , Evariste Njomgue-Fotso , Silvia Bottini
Computational and Structural Biotechnology Journal ( IF 6 ) Pub Date : 2024-03-19 , DOI: 10.1016/j.csbj.2024.03.016 Justine Labory , Evariste Njomgue-Fotso , Silvia Bottini
|
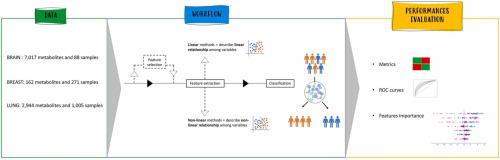
Classification tasks are an open challenge in the field of biomedicine. While several machine-learning techniques exist to accomplish this objective, several peculiarities associated with biomedical data, especially when it comes to omics measurements, prevent their use or good performance achievements. Omics approaches aim to understand a complex biological system through systematic analysis of its content at the molecular level. On the other hand, omics data are heterogeneous, sparse and affected by the classical “curse of dimensionality” problem, i.e. having much fewer observation, samples () than omics features (). Furthermore, a major problem with multi-omics data is the imbalance either at the class or feature level. The objective of this work is to study whether feature extraction and/or feature selection techniques can improve the performances of classification machine-learning algorithms on omics measurements. Among all omics, metabolomics has emerged as a powerful tool in cancer research, facilitating a deeper understanding of the complex metabolic landscape associated with tumorigenesis and tumor progression. Thus, we selected three publicly available metabolomics datasets, and we applied several feature extraction techniques both linear and non-linear, coupled or not with feature selection methods, and evaluated the performances regarding patient classification in the different configurations for the three datasets. We provide general workflow and guidelines on when to use those techniques depending on the characteristics of the data available. To further test the extension of our approach to other omics data, we have included a transcriptomics and a proteomics data. Overall, for all datasets, we showed that applying supervised feature selection improves the performances of feature extraction methods for classification purposes. Scripts used to perform all analyses are available at: https://github.com/Plant-Net/Metabolomic_project/.
中文翻译:

对特征选择和特征提取方法进行基准测试,以提高使用代谢组学生物医学数据进行患者分类的机器学习算法的性能
分类任务是生物医学领域的一个开放挑战。虽然存在多种机器学习技术来实现这一目标,但与生物医学数据相关的一些特性,尤其是在组学测量方面,阻碍了它们的使用或取得良好的性能成就。组学方法旨在通过在分子水平上系统分析其内容来了解复杂的生物系统。另一方面,组学数据是异构的、稀疏的,并且受到经典的“维数灾难”问题的影响,即观察样本()比组学特征()少得多。此外,多组学数据的一个主要问题是类别或特征级别的不平衡。这项工作的目的是研究特征提取和/或特征选择技术是否可以提高分类机器学习算法在组学测量上的性能。在所有组学中,代谢组学已成为癌症研究的强大工具,有助于更深入地了解与肿瘤发生和肿瘤进展相关的复杂代谢景观。因此,我们选择了三个公开的代谢组学数据集,并应用了几种线性和非线性的特征提取技术,无论是否结合特征选择方法,并评估了三个数据集的不同配置中患者分类的性能。我们根据可用数据的特征提供有关何时使用这些技术的一般工作流程和指南。为了进一步测试我们的方法对其他组学数据的扩展,我们纳入了转录组学和蛋白质组学数据。总的来说,对于所有数据集,我们表明应用监督特征选择可以提高用于分类目的的特征提取方法的性能。用于执行所有分析的脚本可从以下网址获取:https://github.com/Plant-Net/Metabolomic_project/。
更新日期:2024-03-19
中文翻译:
对特征选择和特征提取方法进行基准测试,以提高使用代谢组学生物医学数据进行患者分类的机器学习算法的性能
分类任务是生物医学领域的一个开放挑战。虽然存在多种机器学习技术来实现这一目标,但与生物医学数据相关的一些特性,尤其是在组学测量方面,阻碍了它们的使用或取得良好的性能成就。组学方法旨在通过在分子水平上系统分析其内容来了解复杂的生物系统。另一方面,组学数据是异构的、稀疏的,并且受到经典的“维数灾难”问题的影响,即观察样本()比组学特征()少得多。此外,多组学数据的一个主要问题是类别或特征级别的不平衡。这项工作的目的是研究特征提取和/或特征选择技术是否可以提高分类机器学习算法在组学测量上的性能。在所有组学中,代谢组学已成为癌症研究的强大工具,有助于更深入地了解与肿瘤发生和肿瘤进展相关的复杂代谢景观。因此,我们选择了三个公开的代谢组学数据集,并应用了几种线性和非线性的特征提取技术,无论是否结合特征选择方法,并评估了三个数据集的不同配置中患者分类的性能。我们根据可用数据的特征提供有关何时使用这些技术的一般工作流程和指南。为了进一步测试我们的方法对其他组学数据的扩展,我们纳入了转录组学和蛋白质组学数据。总的来说,对于所有数据集,我们表明应用监督特征选择可以提高用于分类目的的特征提取方法的性能。用于执行所有分析的脚本可从以下网址获取:https://github.com/Plant-Net/Metabolomic_project/。


























 京公网安备 11010802027423号
京公网安备 11010802027423号