当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Chemical Bonding and the Role of Node-Induced Electron Confinement
Journal of the American Chemical Society ( IF 15.0 ) Pub Date : 2024-03-26 , DOI: 10.1021/jacs.3c10633 Alistair J. Sterling 1, 2 , Daniel S. Levine 1 , Abdulrahman Aldossary 1 , Martin Head-Gordon 1, 2
Journal of the American Chemical Society ( IF 15.0 ) Pub Date : 2024-03-26 , DOI: 10.1021/jacs.3c10633 Alistair J. Sterling 1, 2 , Daniel S. Levine 1 , Abdulrahman Aldossary 1 , Martin Head-Gordon 1, 2
Affiliation
|
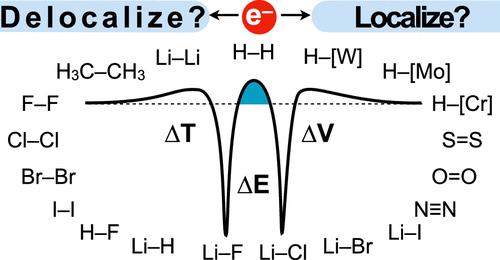
The chemical bond is the cornerstone of chemistry, providing a conceptual framework to understand and predict the behavior of molecules in complex systems. However, the fundamental origin of chemical bonding remains controversial and has been responsible for fierce debate over the past century. Here, we present a unified theory of bonding, using a separation of electron delocalization effects from orbital relaxation to identify three mechanisms [node-induced confinement (typically associated with Pauli repulsion, though more general), orbital contraction, and polarization] that each modulate kinetic energy during bond formation. Through analysis of a series of archetypal bonds, we show that an exquisite balance of energy-lowering delocalizing and localizing effects are dictated simply by atomic electron configurations, nodal structure, and electronegativities. The utility of this unified bonding theory is demonstrated by its application to explain observed trends in bond strengths throughout the periodic table, including main group and transition metal elements.
中文翻译:

化学键合和节点诱导电子限制的作用
化学键是化学的基石,为理解和预测复杂系统中分子的行为提供了概念框架。然而,化学键的基本起源仍然存在争议,并在过去一个世纪引起了激烈的争论。在这里,我们提出了一个统一的成键理论,利用电子离域效应与轨道弛豫的分离来确定三种机制[节点引起的限制(通常与泡利排斥相关,尽管更普遍)、轨道收缩和极化],每种机制都调节键形成过程中的动能。通过对一系列原型键的分析,我们表明,能量降低的离域和局域效应的精确平衡仅由原子电子构型、节点结构和电负性决定。这种统一键合理论的实用性通过其应用来解释整个元素周期表中观察到的键合强度趋势(包括主族和过渡金属元素)来证明。
更新日期:2024-03-26
中文翻译:
化学键合和节点诱导电子限制的作用
化学键是化学的基石,为理解和预测复杂系统中分子的行为提供了概念框架。然而,化学键的基本起源仍然存在争议,并在过去一个世纪引起了激烈的争论。在这里,我们提出了一个统一的成键理论,利用电子离域效应与轨道弛豫的分离来确定三种机制[节点引起的限制(通常与泡利排斥相关,尽管更普遍)、轨道收缩和极化],每种机制都调节键形成过程中的动能。通过对一系列原型键的分析,我们表明,能量降低的离域和局域效应的精确平衡仅由原子电子构型、节点结构和电负性决定。这种统一键合理论的实用性通过其应用来解释整个元素周期表中观察到的键合强度趋势(包括主族和过渡金属元素)来证明。


























 京公网安备 11010802027423号
京公网安备 11010802027423号