当前位置:
X-MOL 学术
›
J. Chin. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Computational modeling of oligothiophenes‐based donor molecules to boost optoelectronic attributes of organic solar cells
Journal of the Chinese Chemical Society ( IF 1.8 ) Pub Date : 2024-03-28 , DOI: 10.1002/jccs.202400033 Adeel Mubarik 1 , Faiza Shafiq 1 , Ammasi Arunkumar 1 , Xue‐Hai Ju 1
Journal of the Chinese Chemical Society ( IF 1.8 ) Pub Date : 2024-03-28 , DOI: 10.1002/jccs.202400033 Adeel Mubarik 1 , Faiza Shafiq 1 , Ammasi Arunkumar 1 , Xue‐Hai Ju 1
Affiliation
|
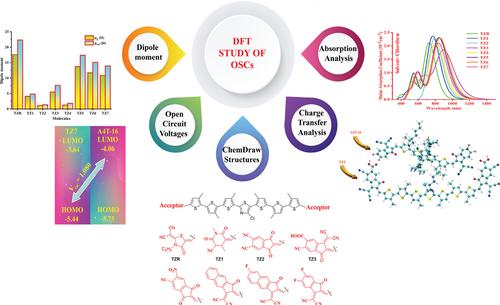
The computational modeling of seven oligothiophene‐based donor molecules (TZ1–TZ7) designed by acceptor modification at the terminal position of the literature molecule (TZR) were discussed for organic solar cells (OSCs). DFT simulations using B3LYP/def2svp levels were performed to study the optoelectronic, and PV properties of TZ1–TZ7. A range of essential aspects for efficient small donor molecules like open circuit voltages (V OC ), excitation energy (E x ), dipole moment (μ), density of state (DOS), absorption maxima (λ max ), transition density matrix (TDM), binding energy (E b ), and frontier molecular orbitals (FMOs) of TZ1–TZ7 and TZR have also been investigated. DOS and FMOs analysis revealed a reduced energy gap (E g ) and effective charge transfer (CT) in the TZ1–TZ7 molecules. The absorption spectra were examined using TD‐DFT. Due to smaller E g , E b , E x , and higher λ max , μ , the TZ1–TZ7 molecules exhibit remarkable optoelectronic properties. The computed V OC (0.969–1.189) and fill factor (0.886–0.897) for TZ1–TZ7 lead to improved power conversion efficiency (PCE) ranging from 14.05% to 17.60%. All compounds are strongly recommended for fabricating efficient OSCs with excellent PV properties. The current work is a step towards environmentally friendly organic PV and will pave the way for future structural engineering research for the efficient material design of OSCs.
中文翻译:

基于低聚噻吩的供体分子的计算模型,以提高有机太阳能电池的光电属性
讨论了有机太阳能电池(OSC)中通过在文献分子(TZR)末端位置进行受体修饰而设计的七个基于低聚噻吩的供体分子(TZ1-TZ7)的计算模型。使用 B3LYP/def2svp 水平进行 DFT 模拟来研究 TZ1–TZ7 的光电和光伏特性。高效小供体分子的一系列重要方面,如开路电压(V 奥克 ), 激发能 (乙 X )、偶极矩 (μ)、态密度 (DOS)、吸收最大值 (λ 最大限度 )、跃迁密度矩阵 (TDM)、结合能 (乙 乙 ),以及 TZ1-TZ7 和 TZR 的前沿分子轨道(FMO)也进行了研究。 DOS 和 FMO 分析显示能隙减小(乙 G )和 TZ1-TZ7 分子中的有效电荷转移(CT)。使用 TD-DFT 检查吸收光谱。由于较小乙 G ,乙 乙 ,乙 X ,以及更高λ 最大限度 ,μ ,TZ1-TZ7分子表现出显着的光电特性。计算出的V 奥克 TZ1–TZ7 的 (0.969–1.189) 和填充因子 (0.886–0.897) 导致功率转换效率 (PCE) 提高,范围从 14.05% 到 17.60%。强烈建议使用所有化合物来制造具有优异光伏性能的高效 OSC。目前的工作是朝着环保有机光伏迈出的一步,将为未来有机太阳能电池高效材料设计的结构工程研究铺平道路。
更新日期:2024-03-28
中文翻译:
基于低聚噻吩的供体分子的计算模型,以提高有机太阳能电池的光电属性
讨论了有机太阳能电池(OSC)中通过在文献分子(TZR)末端位置进行受体修饰而设计的七个基于低聚噻吩的供体分子(TZ1-TZ7)的计算模型。使用 B3LYP/def2svp 水平进行 DFT 模拟来研究 TZ1–TZ7 的光电和光伏特性。高效小供体分子的一系列重要方面,如开路电压(


























 京公网安备 11010802027423号
京公网安备 11010802027423号