当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Balancing Group I Monatomic Ion–Polar Compound Interactions for Condensed Phase Simulation in the Polarizable Drude Force Field
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2024-04-08 , DOI: 10.1021/acs.jctc.3c01380 Yiling Nan 1 , Alexander D. MacKerell 1
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2024-04-08 , DOI: 10.1021/acs.jctc.3c01380 Yiling Nan 1 , Alexander D. MacKerell 1
Affiliation
|
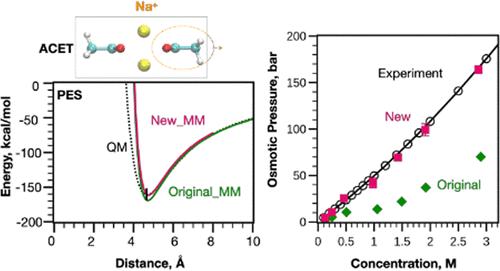
Molecular dynamics (MD) simulations are a commonly used method for investigating molecular behavior at the atomic level. Achieving reliable MD simulation results necessitates the use of an accurate force field. In the present work, we present a protocol to enhance the quality of group 1 monatomic ions (specifically Li+, Na+, K+, Rb+, and Cs+) with respect to their interactions with common polar model compounds in biomolecules in condensed phases in the context of the Drude polarizable force field. Instead of adjusting preexisting individual parameters for ions, model compounds, and water, we employ atom-pair specific Lennard-Jones (LJ) (known as NBFIX in CHARMM) and through-space Thole dipole screening (NBTHOLE) terms to fine-tune the balance of ion–model compound, ion–water, and model compound–water interactions. This involved establishing a protocol for the optimization of NBFIX and NBTHOLE parameters targeting the difference between molecular mechanical (MM) and quantum mechanical (QM) potential energy scans (PES). It is shown that targeting PES involving complexes that include multiple model compounds and/or ions as trimers and tetramers yields parameters that produce condensed phase properties in agreement with experimental data. Validation of this protocol involved the reproduction of experimental thermodynamic benchmarks, including solvation free energies of ions in methanol and N-methylacetamide, osmotic pressures, ionic conductivities, and diffusion coefficients within the condensed phase. These results show the importance of including more complex ion–model compound complexes beyond dimers in the QM target data to account for many-body effects during parameter fitting. The presented parameters represent a significant refinement of the Drude polarizable force field, which will lead to improved accuracy for modeling ion–biomolecular interactions.
中文翻译:

平衡 I 族单原子离子-极性化合物相互作用,用于可极化德鲁德力场中的凝聚相模拟
分子动力学 (MD) 模拟是研究原子水平分子行为的常用方法。获得可靠的 MD 模拟结果需要使用精确的力场。在目前的工作中,我们提出了一个方案,以提高第 1 组单原子离子(特别是 Li +、Na +、K +、Rb +和 Cs +)在凝聚态生物分子中与常见极性模型化合物的相互作用方面的质量。德鲁德极化力场背景下的相位。我们没有调整离子、模型化合物和水预先存在的各个参数,而是采用原子对特定的 Lennard-Jones (LJ)(在 CHARMM 中称为 NBFIX)和全空间 Thole 偶极子筛选 (NBTHOLE) 项来微调离子-模型化合物、离子-水和模型化合物-水相互作用的平衡。这涉及建立一个针对分子力学 (MM) 和量子力学 (QM) 势能扫描 (PES) 之间差异的 NBFIX 和 NBTHOLE 参数优化的协议。结果表明,靶向涉及包含多种模型化合物和/或离子作为三聚体和四聚体的复合物的PES产生的参数产生与实验数据一致的凝聚相特性。该协议的验证涉及实验热力学基准的再现,包括甲醇和N-甲基乙酰胺中离子的溶剂化自由能、渗透压、离子电导率和凝聚相内的扩散系数。这些结果表明,在 QM 目标数据中包含除二聚体之外的更复杂的离子模型化合物复合物,以解释参数拟合期间的多体效应的重要性。所提出的参数代表了德鲁德极化力场的显着改进,这将提高离子-生物分子相互作用建模的准确性。
更新日期:2024-04-08
中文翻译:
平衡 I 族单原子离子-极性化合物相互作用,用于可极化德鲁德力场中的凝聚相模拟
分子动力学 (MD) 模拟是研究原子水平分子行为的常用方法。获得可靠的 MD 模拟结果需要使用精确的力场。在目前的工作中,我们提出了一个方案,以提高第 1 组单原子离子(特别是 Li +、Na +、K +、Rb +和 Cs +)在凝聚态生物分子中与常见极性模型化合物的相互作用方面的质量。德鲁德极化力场背景下的相位。我们没有调整离子、模型化合物和水预先存在的各个参数,而是采用原子对特定的 Lennard-Jones (LJ)(在 CHARMM 中称为 NBFIX)和全空间 Thole 偶极子筛选 (NBTHOLE) 项来微调离子-模型化合物、离子-水和模型化合物-水相互作用的平衡。这涉及建立一个针对分子力学 (MM) 和量子力学 (QM) 势能扫描 (PES) 之间差异的 NBFIX 和 NBTHOLE 参数优化的协议。结果表明,靶向涉及包含多种模型化合物和/或离子作为三聚体和四聚体的复合物的PES产生的参数产生与实验数据一致的凝聚相特性。该协议的验证涉及实验热力学基准的再现,包括甲醇和N-甲基乙酰胺中离子的溶剂化自由能、渗透压、离子电导率和凝聚相内的扩散系数。这些结果表明,在 QM 目标数据中包含除二聚体之外的更复杂的离子模型化合物复合物,以解释参数拟合期间的多体效应的重要性。所提出的参数代表了德鲁德极化力场的显着改进,这将提高离子-生物分子相互作用建模的准确性。


























 京公网安备 11010802027423号
京公网安备 11010802027423号