当前位置:
X-MOL 学术
›
Mol. Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Designing Nickel-based catalysts for the hydrogenation of CO2 under Ambient conditions: A computational study
Molecular Catalysis ( IF 4.6 ) Pub Date : 2024-04-04 , DOI: 10.1016/j.mcat.2024.114089 Anupama Mahato , Akhilesh Mahato , Purnima Singh , Debasis Dhak , Anup Pramanik
Molecular Catalysis ( IF 4.6 ) Pub Date : 2024-04-04 , DOI: 10.1016/j.mcat.2024.114089 Anupama Mahato , Akhilesh Mahato , Purnima Singh , Debasis Dhak , Anup Pramanik
|
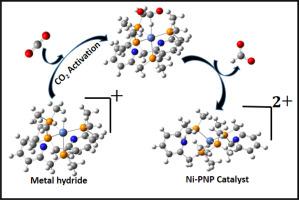
Recent developments, industrialization, and modernization lead to production of more and more pollutants (such as CO: 417.06 ppm) and such rapid growth of CO level results in global warming and the greenhouse effect. So, it is essential to conduct research on converting CO into fuels using carbon-neutral energy. Recent research on molecular catalysts has improved the rates of converting CO to formate. However, these catalysts require extremely high temperatures and pressures and are made of expensive metals like iridium, ruthenium, and rhodium. Herein, DFT-based computational studies have been performed on the catalytic hydrogenation of CO by different Ni(II) complexes in ambient conditions. Using state-of-the-art calculations, we demonstrate how the geometry and spin states of Ni(II) complexes containing different types of ligands affect their catalytic role in CO hydrogenation. It has been reported that hydride transfer is the most crucial step for such kind of hydrogenation reaction. We start with previously synthesised Ni-bis(diphosphine) complexes of type NiP and by extrapolating the concept, we propose a novel kind of Ni-PNP complex with a significantly lower hydride transfer barrier. We also calculate the hydricities of the nickel hydride complexes in order to correlate these thermodynamic parameters with the kinetic barrier of hydride transfer.
中文翻译:

设计用于常温条件下 CO2 加氢的镍基催化剂:计算研究
近年来的发展、工业化和现代化导致产生越来越多的污染物(例如CO:417.06 ppm),CO水平的快速增长导致全球变暖和温室效应。因此,有必要研究利用碳中性能源将二氧化碳转化为燃料。最近对分子催化剂的研究提高了二氧化碳转化为甲酸盐的速率。然而,这些催化剂需要极高的温度和压力,并且由铱、钌和铑等昂贵的金属制成。在此,我们对环境条件下不同 Ni(II) 配合物对 CO 的催化氢化进行了基于 DFT 的计算研究。通过最先进的计算,我们证明了含有不同类型配体的 Ni(II) 配合物的几何形状和自旋态如何影响其在 CO 加氢中的催化作用。据报道,氢化物转移是此类加氢反应中最关键的步骤。我们从之前合成的 NiP 型 Ni-双(二膦)配合物开始,通过推断这一概念,我们提出了一种新型 Ni-PNP 配合物,其氢化物转移势垒显着降低。我们还计算了氢化镍配合物的水度,以便将这些热力学参数与氢化物转移的动力学势垒相关联。
更新日期:2024-04-04
中文翻译:
设计用于常温条件下 CO2 加氢的镍基催化剂:计算研究
近年来的发展、工业化和现代化导致产生越来越多的污染物(例如CO:417.06 ppm),CO水平的快速增长导致全球变暖和温室效应。因此,有必要研究利用碳中性能源将二氧化碳转化为燃料。最近对分子催化剂的研究提高了二氧化碳转化为甲酸盐的速率。然而,这些催化剂需要极高的温度和压力,并且由铱、钌和铑等昂贵的金属制成。在此,我们对环境条件下不同 Ni(II) 配合物对 CO 的催化氢化进行了基于 DFT 的计算研究。通过最先进的计算,我们证明了含有不同类型配体的 Ni(II) 配合物的几何形状和自旋态如何影响其在 CO 加氢中的催化作用。据报道,氢化物转移是此类加氢反应中最关键的步骤。我们从之前合成的 NiP 型 Ni-双(二膦)配合物开始,通过推断这一概念,我们提出了一种新型 Ni-PNP 配合物,其氢化物转移势垒显着降低。我们还计算了氢化镍配合物的水度,以便将这些热力学参数与氢化物转移的动力学势垒相关联。


























 京公网安备 11010802027423号
京公网安备 11010802027423号