Advanced Synthesis & Catalysis ( IF 5.4 ) Pub Date : 2024-04-09 , DOI: 10.1002/adsc.202400174 Lilian Geniller 1 , Marc Taillefer 2 , Eric Clot 1 , Florian Jaroschik 1 , Alexis Prieto 3
|
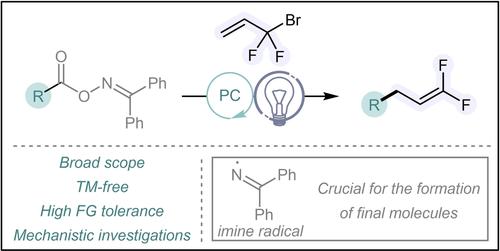
Over the last 15 years, synthetic methods for introducing fluorine atoms into organic compounds have been largely developed, mainly because fluorine incorporation dramatically alters the intrinsically physiological features of organic molecules ranging from increased lipophilicity to higher metabolic stability.1 Consequently, fluorinated molecules have found broad applications in agrochemicals2 and pharmaceuticals.3 Among fluorine-containing groups, gem-difluoroalkenes are extremely interesting as they act both as versatile precursors for building complex fluorine-containing molecules4 and as bioisosteres of carbonyl groups.5 Indeed, the replacement of a carbonyl group by a gem-difluoroalkene moiety can result in a gain of both bioactivity and metabolic stability. This has been demonstrated with the gem-difluoromethylene artemisinin analogue for which the antimalarial activity is higher than for artemisinin (Scheme 1A).6 In this context, considerable efforts have recently been made for developing effective processes for the preparation of gem-difluoroalkenes. Historically, synthesis of such compounds were mainly based on olefination reactions of carbonyl compounds through Julia-Kocienski,7 Horner-Wadsworth-Emmons8 or Wittig processes (Scheme 1B).9 However, the main drawbacks of these approaches are the preparation of “sophisticated” fluorinated precursors and the requirement of basic conditions that might be deleterious for functional groups. The last decade has seen novel and effective strategies relying on the selective defluorofunctionalization of α-CF3-alkenes for building gem-difluoroalkenes (Scheme 1C).10 The key step of these SN2’-type,11 transition-metal-catalyzed,12 or photocatalytic methods13 is the β-elimination of fluoride. However, photocatalytic approaches that require the reduction of the transient carbon radical for triggering the β-fluoride elimination are mainly narrowed to the defluorofunctionalization of α-CF3-styrenes, reducing their broader applicability (Scheme 1C). More recently, an elegant photocatalytic procedure has been reported for the exclusive construction of five-membered rings bearing gem-difluoroalkenyl or monofluoroalkenyl patterns by the use of alkynes and mono- or difluorodibromoalkanes (Scheme 1D).14 Mechanistically, this procedure is in sharp contrast with other photocatalyzed methods as the key step is believed to be a β-scission that would form gem-difluoroalkenes along with the release of a bromine radical. Concomitantly to this work, we have demonstrated that light-induced radical β-bromo or iodo fragmentation can be key to achieving hydrohalogenation of unsaturated compounds under visible-light irradiation.15 In this context and given our recent interest for fluorination reactions,16 we envisaged to explore the possibility of synthesizing gem-difluoroalkenes by the use of readily available oxime ester derivatives and 1-bromo-1,1-difluoroprop-2-ene (BDFP). Indeed, such oximes have recently been shown to be efficient radical precursors for achieving a large panel of organic reactions.17 They can simply be activated via energy transfer (EnT) providing the concomitant formation of carbon and imine radicals through N−O bond cleavage and a decarboxylative process. Those radicals can thereafter be involved in various organic reactions. It has already been shown that in the presence of alkenes, carbon and imine radicals afford essentially 1,2-difunctionalized compounds, which arise from the successive addition of carbon radicals onto alkenes, followed by the radical-radical couplings (RRC) of these intermediates with the imine radical (Scheme 1E).18

Examples of bioactive gem-difluoroalkenes and their preparations.
In our case, the radical intermediates, generated from the radical functionalization of the BDFP, would have the possibility to be part in either the classical RRC (red arrows) or the β-bromo fragmentation (green arrows) (Scheme 1F). However, we hypothesized that the radical intermediate would be more prone to undergo the faster monomolecular β-scission than the bimolecular RRC (Scheme 1F). We herein present the successful development of the described strategy that gives rise to a large variety of functionalized gem-difluoroalkenes, going beyond styrene derivatives. Moreover, experimental and computational mechanistic studies allowed us to demonstrate the crucial role of imine radical either as a bromine radical scavenger or as an XAT promoter for the reaction.
To test the feasibility of our photochemical plan, we selected the diphenylmethanone O-(4-(4-methoxyphenyl)butanoyl)oxime 1 a as model substrate to react with BDFP (2). After optimization of photocatalyst (PC), solvent, dilution, light wavelength and stoichiometric ratio, the reaction was found to be the most effective when 3 equivalents of oxime 1 a, 1 equivalent of BDFP, and 10 mol% of thioxanthone (TX) as PC were used in AcOEt under 390 nm irradiation. In these conditions, the gem-difluoroalkene 3 a was obtained in an excellent isolated yield of 93% along with the compound 4 a, isolated in a 71% yield. (Table 1, entry 1). Despite significant efforts, we could not find any conditions preventing the formation of 4 a, and this byproduct explains the necessity of using 3 equivalents of oxime 1 a. The use of Ir- or Ru-based photocatalysts in combination with blue light was less productive, with formation of 3 a in lower yield or complete absence of 3 a (Table 1, entries 2–3).
|
|||
Entry |
Deviation from standard conditions |
Yields (%)a |
|
|---|---|---|---|
3 a |
4 a |
||
1 |
None |
Quant (93)b |
71b |
2 |
(Ir[dF(CF3)ppy]2(dtbpy))PF6 (1 mol%)c |
70 |
37 |
3 |
[Ru(ppy)3Cl2].6H2O (3 mol%)c |
0 |
0 |
4 |
TX (5 mol%) |
85 |
48 |
5 |
Acetone |
quant |
83 |
6 |
DCM |
94 |
76 |
7 |
benzene |
84 |
68 |
8 |
1 a (2 equiv.) |
43 |
30 |
9 |
1 a (1 equiv.), 2 (2 equiv.) |
20 |
14 |
10 |
w/o TX |
20 |
5 |
11 |
In the dark |
N.R. |
|

- Reaction conditions: 2 (0.3 mmol), 1 a (0.9 mmol) in AcOEt (1 mL). [a] Yields were determined by 1H NMR using the trichloroethylene as an internal standard. [b] Isolated yield. [c] Under 450 nm irradiation. TX=Thioxanthone. N.R.=No reaction.
The decrease of the catalytic loading of TX to 5 mol% caused a slight drop of yield (Table 1, entry 4). The use of other solvents did not affect reaction outcomes providing 3 a in similar yields compared to AcOEt (Table 1, entries 5–7). When a lower amount of oxime 1 a was used, the reaction provided 3 a, albeit in a moderate yield of 43% (Table 1, entry 8). A similar observation was made when the reaction was set up with 2 equivalents of BDFP and 1 equivalent of oxime 1 a (Table 1, entry 9). Finally, control experiments showed that both the photocatalyst and the light were essential for the reaction to proceed efficiently (Table 1, entries 10–11).
With the optimized reaction conditions in hand, we then explored the scope and the limitations of the reaction (Scheme 2). The reaction was first studied with oxime derivatives 1 that would give rise to alkyl radicals upon photocatalysis. The reaction using a wide variety of primary alkyl oximes (1 a–1 o) provided successfully the expected gem-difluoromethylene 3 a–3 o in good to excellent yields. Remarkably, the presence of various functional groups, such as halides, esters, alkenes, alkynes, phosphonates, nitro, native hydroxyl, or (thio)ethers did not affect the reaction outcome, demonstrating the good functional group tolerance of the reaction (Scheme 2). We could also demonstrate the usefulness of the reaction by securing a gram-scale experiment. Pleasingly, even on a 10 mmol scale, the reaction provided the expected product 3 c in a similar yield compared to the 0.5 mmol scale. Then, the reaction was studied with secondary and tertiary alkyl oximes (1 p–1 x) (Scheme 2). Overall, the reaction afforded the corresponding gem-difluoroalkene compounds in moderate to good yields (3 p–3 x). It should be noticed that the reaction with the adamantyl oxime derivative 1 v, led also to the formation of adamantane. The formation of a defunctionalized compound was only observed in this case. We were pleased to see that the use of 2 equivalents of oxime 1 v and the reduction of the reaction time to 12 h, avoided the formation of adamantane. We then turned our attention to the use of aryl oxime esters. Unfortunately, the reaction was found rather ineffective with oximes, such as 1 y, delivering the corresponding compound 3 y in poor yield. Then, to demonstrate that our method constitutes a useful approach for the synthesis of complex gem-difluoroalkenes, a large variety of oximes derived from natural and drug molecules have been employed (Scheme 2). Simple natural molecules, such as stearic and oleic acid derivatives gave final products 3 z and 3 aa in nearly quantitative yields. Then, the reaction was explored with oxime derivatives stemming from natural isoleucine and tryptophan amino acids. Pleasingly, the reaction offered the formation of substrates 3 ab and 3 ac in 54% moderate yields. Steroid molecules derived from dehydrocholic acid and lithocholic acid were also found to be good candidates in the reaction providing the corresponding molecules 3 ad–3 ae in good yields. The gem-difluoroalkene 3 af, derived from gemfibrozil, was also obtained in a nearly quantitative yield. Finally, the reaction was investigated using other CF2Br-alkenes bearing an additional substituent in the 2-position. While the reaction with alkyl-substituted CF2Br-alkene 2 b gave rise to the product 3 ag in a good yield of 63%, in the case of the aryl-substituted substrate 2 c the gem-difluorostyrene 3 ah was obtained in a moderate yield of 35% (Scheme 2). These results highlight that our methodology is complementary to the photocatalytic defluorofunctionalization strategy. Indeed, even though the synthesis of gem-difluorostyrenes seems to be less effective with the current approach, the synthesis of di-alkyl substituted difluoromethylenes would be hardly efficient using photocatalytic defluorofunctionalization strategies.13

Scope of the reaction. Reaction conditions: 2 (0.5 mmol), 1 (1.5 mmol) in AcOEt (1.7 mL). Yields refer to isolated products.[a] 1H NMR yield is given.[b] Reaction performed on 10 mmol scale.[c] Compound highly volatile.[d] 2 equivalents of the corresponding oxime were used and the reaction time was reduced to 12 h.[e] Reaction performed in 5 mL.
Next, we focused our attention to the mechanistic elucidation of the newly developed procedure. Several control experiments have been performed along with DFT calculations. First, we decided to conduct the reaction in the presence of the radical scavenger 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO, 3 equiv.) (Scheme 3A). In those conditions, only traces of the compounds 3 c and 4 c could be observed by NMR analyses. However, we could detect by MS analysis the formation of the adduct TEMPO-CH2CH2Ph.

Mechanistic experiments.
When citronellic acid derived oxime was used, the reaction led to the formation of the ring-closing product 3 ai in an excellent yield (Scheme 3B). Both experiments support the proposition that carbon radicals are generated under these reaction conditions. Based on these results we also attempted to generate the carbon radicals from a peroxide precursor under thermal conditions, but no product formation was observed (see SI). Thereafter, we tried to understand and explain the formation of compounds 4. Considering that the putative radical β-bromo fragmentation would release a bromine radical and that compounds 4 were unlikely to be formed via a radical-radical coupling between bromine radical and the generated carbon radicals, we thought that compounds 4 might result from an XAT between those carbon radicals and an in-situ formed bromide source. Indeed, the generation of bromine radicals might either lead to the formation of Br2 or to the N-bromo-1,1-diphenylmethanimine (5) (Scheme 3C). We therefore synthesized 5 and employed it in the normal reaction conditions. Compound 4 c was obtained in a good yield of 71% along with 10% of styrene. On the other hand, the reaction using Br2 as bromine source did not allow the formation of 4 c. These results tend to prove that the N-bromoimine 5 is likely formed during the reaction and acts as a bromine source.
To shed more light on the observed reactivity, DFT calculations (PBE0-D3) have been carried out (see ESI for the Computational Details). As the reaction was successful with simple alkyl chains (see 3 z), the diphenylmethanone methyloxime 1 aj was selected as model substrate for DFT calculations. Geometry optimizations of TX and 1 aj in a triplet state allowed to position energetically these excited states with respect to the corresponding singlet ground states (ΔGS/T=58.9 kcal.mol−1, TX; ΔGS/T=45.3 kcal.mol−1, 1 aj).17e Efficient energy transfer (EnT) from the triplet excited state of TX to the singlet ground state of 1 aj promotes the oxime in its first triplet excited state T1(1 aj). The latter is the starting point for further reactivity (Scheme 4, see ESI for details about calculations of excited states).

Computed reaction mechanism (ΔG, kcal.mol−1) for the formation of the methyl radical from the excited triplet state of the oxime and its further reactivity.
The first step on the reaction pathway is associated with the cleavage of the N−O bond in T1(1 aj) through TS1 (N−O=1.363 Å, T1(1 aj); N…O=1.760 Å, TS1). The activation barrier is computed to be low (ΔGǂ=8.0 kcal.mol−1) and the reaction forming the radicals Me−CO2⋅ (A) and ⋅N=CPh2 (B) is exoergic (ΔG=−13.5 kcal.mol−1). Decarboxylation of A to generate ⋅CH3 (C) and CO2 is, as expected, computed to be easy (ΔGǂ=5.5 kcal.mol−1) and exoergic (ΔG=−7.6 kcal.mol−1). Further reaction of the methyl radical C with the alkene H2C=CH(CF2Br) is also computed to be easy (ΔGǂ=10.7 kcal.mol−1) and strongly exoergic (ΔG=−23.8 kcal.mol−1) as a new C−C bond is formed. Once the oxime 1 aj is photoexcited, the sequence of steps leading to the alkene-derived functionalized radical D is favored both kinetically and thermodynamically (Scheme 4). Formation of D is associated to a slight elongation of the C−Br bond from 1.97 Å in H2C=CH(CF2Br) to 2.03 Å in D.
From the intermediate D three pathways might be considered; 1/a β-bromo scission,14, 15 2/a bromide abstraction (XAT), 3/a bimolecular RRC reaction (Scheme 5). While the first two possibilities would allow the formation of the expected gem-difluoroalkene 3 aj, the last option would lead to the formation of the 1,2-difunctionalized compound F. Calculations showed that the β-scission step releasing Br⋅ from D is slightly endoergic (ΔG=9.5 kcal.mol−1, Scheme 5).19 Despite numerous attempts, no transition state structure for the transfer of Br⋅ from D to ⋅N=CPh2 could be located on the potential energy surface (PES). However, the thermodynamic driving force associated to the formation of the N−Br bond in 5 (ΔG=−36.5 kcal.mol−1) strongly stabilizes the product 3 aj with respect to D (Scheme 5). These results point out the crucial role of the imine radical B for the formation of 3 aj. Considering the endoergic β-scission process, the radical imine B would act as a bromide radical scavenger, allowing the displacement of the reaction toward the formation of 3 aj. On the other hand, we could not exclude that the imine radical B might also act as an XAT promoter performing the direct bromide abstraction on intermediate D. Both scenarios would in fine lead to the formation of N-bromoimine (5, Br−N=CPh2). The latter might be involved in a bromine atom transfer with ⋅CH3, computed to be easy kinetically (ΔGǂ=7.9 kcal.mol−1) and favored thermodynamically (ΔG=−30.3 kcal.mol−1), explaining the formation of 4 aj (CH3Br, Scheme 5). This agrees with the experimental observations of formation of both 3 a and 4 a and the need of 3 equiv. of 1 a to perform the reaction (Table 1).

Computed competition reactions (ΔG, kcal.mol−1) starting from the brominated radical D.
Finally, the bimolecular RRC reaction between D and ⋅N=CPh2 to form 1F was calculated and, as expected, was shown to be strongly exergonic (ΔG=−44.3 kcal.mol−1) and thermodynamically favored over the formation of the experimentally observed product 3 aj (Scheme 5). However, neither NMR analyses of crude reactions nor NMR monitoring of the reaction allowed us to observe the formation of 1F. As the whole pathway shown in Scheme 4 relies on the photogenerated cleavage of the N−O bond in the oxime, we reasoned that reversibility in the formation of 1F could potentially be obtained upon photoexcitation. The triplet state, 3F, for the product of bimolecular RRC is computed to lie at ΔG=42.3 kcal.mol−1 above 1F. From 3F, a transition state structure, 3TS5, associated to C−N bond cleavage, could be located with ΔGǂ=20.0 kcal.mol−1. As the energy difference between 1F and 3F (ΔG=42.3 kcal.mol−1) is comparable to that computed between S0(1 aj) and T1(1 aj) (ΔG=45.3 kcal.mol−1), energy transfer from the photoexcited TX to 1F could be efficient. This supports a potential reversible pathway that would eventually lead to the exclusive formation of the olefin 3 aj through the regeneration of D evolving thereafter following β-bromo fragmentation or bromide abstraction pathways. However, by virtue of the principle of micro reversibility, the transition state, 3TS5, to form the C−N bond in 3F is higher in energy than the energy required to fully dissociate Br⋅ from D (18 vs 9.5 kcal.mol−1). Therefore, once D is formed, the presence of the persistent radical ⋅N=CPh2 triggers the system toward exclusive formation of the gem-difluoroalkene 3 aj. The aforementioned reversibility was then studied experimentally by specifically preparing the substrate 7 (Scheme 6).20 However, submitting 7 to the experimental conditions used to perform the reaction shown in Scheme 1, only gave very poor conversion (<10%) and no gem-difluoroalkene formation could be observed, discarding any reversibility process in this reaction.

Study of reversibility.
In conclusion, we have established a new and general protocol for the synthesis of a large variety of alkyl-substituted gem-difluoroalkenes that includes complex molecules and drug derivatives by the use of readily available oxime esters and BDFP. This method overcomes previous limitations encountered in reported photoredox syntheses of gem-difluoroalkenes, which are limited to the access of gem-difluorostyrenes. DFT calculations point out the crucial role of imine radical for the reaction efficiency. However, even though the role of imine radical is not clearly assigned, the latter might act as either a bromine radical scavenger or a XAT promoter in the current transformation. Future work will focus on exploiting this new role of imine radical in photocatalytic processes, and explore imine substrates as carbon radical precursors.
Experimental Section
Preparation of gem-Difluoroalkenes 3; General Procedure: In a vial, 3-bromo-3,3-difluoroprop-1-ene (51 μL, 10 mmol, 1 equiv.) was added to a solution of the corresponding oxime ester (1.5 mmol, 3 equiv.) and thioxanthone (10.6 mg, 0.05 mmol, 10 mol %) in AcOEt (1.7 mL). The reaction mixture was degassed via a freeze pump thaw procedure, and then placed in the EvoluChemTM PhotoRedOx reactor fitted with the Kessil PR160 L – 390 nm light. The reaction was stirred under light irradiation at room temperature for 18 h and then concentrated in vacuo. Then the residue was purified by silica gel column chromatography.
Acknowledgments
This research was supported financially by a grant to L.G. from the French Ministry of Higher Education and Research delivered by the Doctoral school (ED 459) and the ICGM. We thank the Centre National de la Recherche Scientifique (CNRS), the Ecole Nationale Supérieure de Chimie de Montpellier (ENSCM), the Université de Montpellier for financial support. The authors also thank the Balard Platform Analysis and Characterization (PAC Balard) facilities for technical support. The French Fluorine Network (GIS fluor) is also acknowledged.
中文翻译:
使用肟酯光催化形成偕二氟烯烃
在这篇手稿中,建立了一种使用现成的肟酯和1-溴-1,1-二氟丙-2-烯(BDFP)合成烷基取代的偕二氟烯烃的通用光催化方法。这种涉及自由基消除溴的策略提供了获得多种增值氟化分子的途径。温和的反应条件与许多官能团兼容,包括复杂的天然产物或药物分子。实验和理论机理研究表明,该过程的效率取决于亚胺自由基的关键作用,亚胺自由基是在 EnT 光激发和肟酯脱羧后形成的。事实上,亚胺自由基在该反应中要么充当溴自由基清除剂,要么充当 XAT 促进剂。


























 京公网安备 11010802027423号
京公网安备 11010802027423号