当前位置:
X-MOL 学术
›
Mol. Pharmaceutics
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Transfer of ANS-Like Drugs from Micellar Drug Delivery Systems to Albumin Is Highly Favorable and Protected from Competition with Surfactant by “Reserved” Binding Sites
Molecular Pharmaceutics ( IF 4.9 ) Pub Date : 2024-04-16 , DOI: 10.1021/acs.molpharmaceut.3c00875 Iulia Carabadjac 1 , Leonie C. Vormittag 1 , Thomas Muszer 1 , Jakob Wuth 1 , Maximilian H. Ulbrich 2, 3 , Heiko Heerklotz 1, 3, 4
Molecular Pharmaceutics ( IF 4.9 ) Pub Date : 2024-04-16 , DOI: 10.1021/acs.molpharmaceut.3c00875 Iulia Carabadjac 1 , Leonie C. Vormittag 1 , Thomas Muszer 1 , Jakob Wuth 1 , Maximilian H. Ulbrich 2, 3 , Heiko Heerklotz 1, 3, 4
Affiliation
|
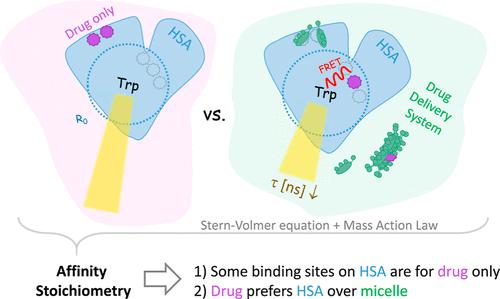
Micellar drug delivery systems (MDDS) for the intravenous administration of poorly soluble drugs have great advantages over alternative formulations in terms of the safety of their excipients, storage stability, and straightforward production. A classic example is mixed micelles of glycocholate (GC) and lecithin, both endogenous substances in human blood. What limits the use of MDDS is the complexity of the transitions after injection. In particular, as the MDDS disintegrate partially or completely after injection, the drug has to be transferred safely to endogenous carriers in the blood, such as human serum albumin (HSA). If this transfer is compromised, the drug might precipitate─a process that needs to be excluded under all circumstances. The key question of this paper is whether the high local concentration of GC at the moment and site of MDDS dissolution might transiently saturate HSA binding sites and, hence, endanger quick drug transfer. To address this question, we have used a new approach, which is time-resolved fluorescence spectroscopy of the single tryptophan in HSA, Trp-214, to characterize the competitive binding of GC and the drug substitute anilinonaphthalenesulfonate (ANS) to HSA. Time-resolved fluorescence of Trp-214 showed important advantages over established methods for tackling this problem. ANS has been the standard “model drug” to study albumin binding for decades, given its structural similarity to the class of naphthalene-containing acidic drugs and the fact that it is displaced from HSA by numerous drugs (which presumably bind to the same sites). Our complex global fit uses the critical approximation that the average lifetimes behave similarly to a single lifetime, but the resulting errors are found to be moderate and the results provide a convincing explanation of the, at first glance, counterintuitive behavior. Accordingly, and largely in line with the literature, we observed two types of sites binding ANS at HSA: 3 type A, rather peripheral, and 2 type B, likely more central sites. The latter quench Trp-214 by Förster Resonance Energy Transfer (FRET) with a rate constant of ≈0.4 ns–1 per ANS. Adding millimolar concentrations of GC displaces ANS from the A sites but not from B sites. At incomplete ANS saturation, this causes a GC-induced translocation of ANS from A to the more FRET-active B sites. This leads to the apparent paradox that the partial displacement of ANS from HSA increases its quenching effect on Trp-214. The most important conclusion is that (ANS-like) drugs cannot be displaced from the type-B sites, and consequently, drug transfer to these sites is not impaired by competitive binding of GC in the vicinity of a dissolving micelle. The second conclusion is that for unbound GC above the CMC (9 mM), ANS equilibrates between HSA and GC micelles but with a strong preference for free sites on HSA. That means that even persisting micelles would lose their cargo readily once exposed to HSA. For all MDDS sharing this property, targeted drug delivery approaches involving them as the nanocarrier would be pointless.
中文翻译:

ANS 类药物从胶束药物递送系统转移到白蛋白是非常有利的,并且通过“保留”的结合位点免受与表面活性剂的竞争
用于静脉注射难溶性药物的胶束给药系统(MDDS)在赋形剂的安全性、储存稳定性和直接生产方面比其他制剂具有巨大的优势。一个典型的例子是甘胆酸 (GC) 和卵磷脂的混合胶束,这两种物质都是人体血液中的内源性物质。 MDDS 的使用受到限制的是注射后转变的复杂性。特别是,由于 MDDS 在注射后部分或完全崩解,因此药物必须安全地转移到血液中的内源性载体,例如人血清白蛋白 (HSA)。如果这种转移受到损害,药物可能会沉淀——在任何情况下都需要排除这一过程。本文的关键问题是 MDDS 溶解时和部位的 GC 局部高浓度是否可能暂时使 HSA 结合位点饱和,从而危及药物的快速转移。为了解决这个问题,我们使用了一种新方法,即 HSA 中单一色氨酸 Trp-214 的时间分辨荧光光谱,来表征 GC 和药物替代品苯胺萘磺酸盐 (ANS) 与 HSA 的竞争性结合。 Trp-214 的时间分辨荧光显示出与解决该问题的现有方法相比的重要优势。 ANS 几十年来一直是研究白蛋白结合的标准“模型药物”,因为它的结构与含萘酸性药物类别相似,而且它被多种药物(推测与相同位点结合)从 HSA 中取代。 。我们复杂的全局拟合使用临界近似,即平均寿命的行为与单个寿命相似,但发现所产生的误差是适度的,并且结果为乍一看违反直觉的行为提供了令人信服的解释。因此,与文献基本一致,我们观察到 HSA 上有两种类型的 ANS 结合位点:3 个 A 型位点,相当外围,以及 2 个 B 型位点,可能更中心。后者通过福斯特共振能量转移 (FRET) 淬灭 Trp-214,速率常数为 ≈0.4 ns –1根据 ANS。添加毫摩尔浓度的 GC 会取代 A 位点上的 ANS,但不会取代 B 位点上的 ANS。在 ANS 不完全饱和时,这会导致 GC 诱导的 ANS 从 A 易位到 FRET 活性更高的 B 位点。这导致了一个明显的悖论,即 ANS 从 HSA 的部分置换增加了其对 Trp-214 的猝灭作用。最重要的结论是(ANS 样)药物不能从 B 型位点转移,因此,药物向这些位点的转移不会因溶解胶束附近 GC 的竞争性结合而受到损害。第二个结论是,对于高于 CMC (9 mM) 的未结合 GC,ANS 在 HSA 和 GC 胶束之间达到平衡,但强烈偏爱 HSA 上的自由位点。这意味着,即使是持久存在的胶束,一旦暴露于 HSA,也会轻易失去其所含的物质。对于所有具有这种特性的 MDDS,将它们作为纳米载体的靶向药物递送方法将毫无意义。
更新日期:2024-04-16
中文翻译:
ANS 类药物从胶束药物递送系统转移到白蛋白是非常有利的,并且通过“保留”的结合位点免受与表面活性剂的竞争
用于静脉注射难溶性药物的胶束给药系统(MDDS)在赋形剂的安全性、储存稳定性和直接生产方面比其他制剂具有巨大的优势。一个典型的例子是甘胆酸 (GC) 和卵磷脂的混合胶束,这两种物质都是人体血液中的内源性物质。 MDDS 的使用受到限制的是注射后转变的复杂性。特别是,由于 MDDS 在注射后部分或完全崩解,因此药物必须安全地转移到血液中的内源性载体,例如人血清白蛋白 (HSA)。如果这种转移受到损害,药物可能会沉淀——在任何情况下都需要排除这一过程。本文的关键问题是 MDDS 溶解时和部位的 GC 局部高浓度是否可能暂时使 HSA 结合位点饱和,从而危及药物的快速转移。为了解决这个问题,我们使用了一种新方法,即 HSA 中单一色氨酸 Trp-214 的时间分辨荧光光谱,来表征 GC 和药物替代品苯胺萘磺酸盐 (ANS) 与 HSA 的竞争性结合。 Trp-214 的时间分辨荧光显示出与解决该问题的现有方法相比的重要优势。 ANS 几十年来一直是研究白蛋白结合的标准“模型药物”,因为它的结构与含萘酸性药物类别相似,而且它被多种药物(推测与相同位点结合)从 HSA 中取代。 。我们复杂的全局拟合使用临界近似,即平均寿命的行为与单个寿命相似,但发现所产生的误差是适度的,并且结果为乍一看违反直觉的行为提供了令人信服的解释。因此,与文献基本一致,我们观察到 HSA 上有两种类型的 ANS 结合位点:3 个 A 型位点,相当外围,以及 2 个 B 型位点,可能更中心。后者通过福斯特共振能量转移 (FRET) 淬灭 Trp-214,速率常数为 ≈0.4 ns –1根据 ANS。添加毫摩尔浓度的 GC 会取代 A 位点上的 ANS,但不会取代 B 位点上的 ANS。在 ANS 不完全饱和时,这会导致 GC 诱导的 ANS 从 A 易位到 FRET 活性更高的 B 位点。这导致了一个明显的悖论,即 ANS 从 HSA 的部分置换增加了其对 Trp-214 的猝灭作用。最重要的结论是(ANS 样)药物不能从 B 型位点转移,因此,药物向这些位点的转移不会因溶解胶束附近 GC 的竞争性结合而受到损害。第二个结论是,对于高于 CMC (9 mM) 的未结合 GC,ANS 在 HSA 和 GC 胶束之间达到平衡,但强烈偏爱 HSA 上的自由位点。这意味着,即使是持久存在的胶束,一旦暴露于 HSA,也会轻易失去其所含的物质。对于所有具有这种特性的 MDDS,将它们作为纳米载体的靶向药物递送方法将毫无意义。


























 京公网安备 11010802027423号
京公网安备 11010802027423号