当前位置:
X-MOL 学术
›
Mol. Biosyst.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
A computational method using differential gene expression to predict altered metabolism of multicellular organisms
Molecular BioSystems Pub Date : 2017-09-22 00:00:00 , DOI: 10.1039/c7mb00462a Lvxing Zhu 1, 2, 3, 4 , Haoran Zheng 1, 2, 3, 4, 5 , Xinying Hu 1, 2, 3, 4 , Yang Xu 1, 2, 3, 4
Molecular BioSystems Pub Date : 2017-09-22 00:00:00 , DOI: 10.1039/c7mb00462a Lvxing Zhu 1, 2, 3, 4 , Haoran Zheng 1, 2, 3, 4, 5 , Xinying Hu 1, 2, 3, 4 , Yang Xu 1, 2, 3, 4
Affiliation
|
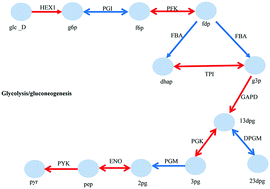
Altered metabolism is often identified as a cause or an effect of physiology and pathogenesis. But it is difficult to predict the metabolic flux distributions of multicellular organisms due to the lack of an explicit metabolic objective function. Here we present a computational method which can successfully describe the differences in metabolism between two different conditions on a large scale. By integrating gene expression data with an existing comprehensive reconstruction of the global human metabolic network, we qualitatively predicted significantly differential fluxes without prior knowledge or the rate of metabolite uptake and secretion. Therefore, this method can be applied for both microorganisms and multicellular organisms. Different from traditional enrichment analysis methods and constraint-based models, we consider conditions and interactions within the metabolic network simultaneously. To apply the proposed method, we predicted altered fluxes for E. coli strains and clear cell renal cell carcinoma, while the E. coli strains are growing aerobically in a chemostat with different dilution rates and clear cell renal cell carcinoma is compared with normal kidney cells. Then we map the significantly differential reactions to metabolic subsystems defined in the original metabolic network for ccRCC to observe the altered metabolism. In contrast with existing studies, our results show a high accuracy of the E. coli experiment and a more reasonable prediction of the ccRCC experiment. The method presented here provides a computational approach for the genome-wide study of altered metabolism under pairs of conditions for both microorganisms and multicellular organisms.
中文翻译:

一种使用差异基因表达预测多细胞生物代谢改变的计算方法
代谢改变通常被鉴定为生理学和发病机理的原因或影响。但是由于缺乏明确的代谢目标功能,很难预测多细胞生物的代谢通量分布。在这里,我们提出了一种计算方法,可以成功地大规模描述两种不同条件之间的代谢差异。通过将基因表达数据与全球人类代谢网络的现有全面重建相结合,我们可以定性地预测明显的差异通量,而无需先验知识或代谢物吸收和分泌的速率。因此,该方法可以应用于微生物和多细胞生物。与传统的浓缩分析方法和基于约束的模型不同,我们同时考虑代谢网络中的条件和相互作用。为了应用提出的方法,我们预测了大肠杆菌菌株和透明细胞肾细胞癌,而大肠杆菌菌株在具有不同稀释率的恒化器中需氧生长,并且将透明细胞肾细胞癌与正常肾细胞进行比较。然后,我们将显着不同的反应映射到ccRCC原始代谢网络中定义的代谢子系统,以观察变化的代谢。与现有研究相反,我们的结果显示了大肠杆菌实验的高精度和ccRCC实验的更合理的预测。本文介绍的方法为微生物和多细胞生物对在成对条件下代谢变化的全基因组研究提供了一种计算方法。
更新日期:2017-10-25
中文翻译:
一种使用差异基因表达预测多细胞生物代谢改变的计算方法
代谢改变通常被鉴定为生理学和发病机理的原因或影响。但是由于缺乏明确的代谢目标功能,很难预测多细胞生物的代谢通量分布。在这里,我们提出了一种计算方法,可以成功地大规模描述两种不同条件之间的代谢差异。通过将基因表达数据与全球人类代谢网络的现有全面重建相结合,我们可以定性地预测明显的差异通量,而无需先验知识或代谢物吸收和分泌的速率。因此,该方法可以应用于微生物和多细胞生物。与传统的浓缩分析方法和基于约束的模型不同,我们同时考虑代谢网络中的条件和相互作用。为了应用提出的方法,我们预测了大肠杆菌菌株和透明细胞肾细胞癌,而大肠杆菌菌株在具有不同稀释率的恒化器中需氧生长,并且将透明细胞肾细胞癌与正常肾细胞进行比较。然后,我们将显着不同的反应映射到ccRCC原始代谢网络中定义的代谢子系统,以观察变化的代谢。与现有研究相反,我们的结果显示了大肠杆菌实验的高精度和ccRCC实验的更合理的预测。本文介绍的方法为微生物和多细胞生物对在成对条件下代谢变化的全基因组研究提供了一种计算方法。


























 京公网安备 11010802027423号
京公网安备 11010802027423号