当前位置:
X-MOL 学术
›
Mol. Biosyst.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Evaluation and optimization of reduction and alkylation methods to maximize peptide identification with MS-based proteomics
Molecular BioSystems Pub Date : 2017-09-20 00:00:00 , DOI: 10.1039/c7mb00393e Suttipong Suttapitugsakul 1, 2, 3, 4 , Haopeng Xiao 1, 2, 3, 4 , Johanna Smeekens 1, 2, 3, 4 , Ronghu Wu 1, 2, 3, 4
Molecular BioSystems Pub Date : 2017-09-20 00:00:00 , DOI: 10.1039/c7mb00393e Suttipong Suttapitugsakul 1, 2, 3, 4 , Haopeng Xiao 1, 2, 3, 4 , Johanna Smeekens 1, 2, 3, 4 , Ronghu Wu 1, 2, 3, 4
Affiliation
|
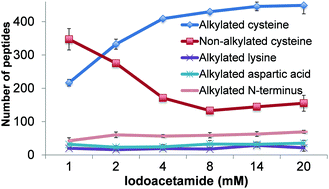
Mass spectrometry (MS) has become an increasingly important technique to analyze proteins. In popular bottom-up MS-based proteomics, reduction and alkylation are routine steps to facilitate peptide identification. However, incomplete reactions and side reactions may occur, which compromise the experimental results. In this work, we systematically evaluated the reduction step with commonly used reagents, i.e., dithiothreitol, 2-mercaptoethanol, tris(2-carboxyethyl)phosphine, or tris(3-hydroxypropyl)phosphine, and alkylation with iodoacetamide, acrylamide, N-ethylmaleimide, or 4-vinylpyridine. By using digested peptides from a yeast whole-cell lysate, the number of proteins and peptides identified were very similar using four different reducing reagents. The results from four alkylating reagents, however, were dramatically different with iodoacetamide giving the highest number of peptides with alkylated cysteine and the lowest number of peptides with incomplete cysteine alkylation and side reactions. Alkylation conditions with iodoacetamide were further optimized. To identify more peptides with cysteine, thiopropyl-sepharose 6B resins were used to enrich them, and the optimal conditions were employed for the reduction and alkylation. The enrichment resulted in over three times more cysteine-containing peptides than without enrichment. Systematic evaluation of the reduction and alkylation with different reagents can aid in a better design of bottom-up proteomic experiments.
中文翻译:

评估和优化还原和烷基化方法,以最大程度地利用基于MS的蛋白质组学鉴定多肽
质谱(MS)已成为分析蛋白质的越来越重要的技术。在流行的自下而上的基于MS的蛋白质组学中,还原和烷基化是促进肽鉴定的常规步骤。但是,可能会发生不完全反应和副反应,从而损害了实验结果。在这项工作中,我们系统地评估了常用试剂即二硫苏糖醇,2-巯基乙醇,三(2-羧乙基)膦或三(3-羟丙基)膦的还原步骤,以及碘乙酰胺,丙烯酰胺,N的烷基化-乙基马来酰亚胺或4-乙烯基吡啶。通过使用酵母全细胞裂解液中消化的肽,使用四种不同的还原剂,鉴定出的蛋白质和肽的数量非常相似。然而,四种烷基化试剂的结果与碘乙酰胺截然不同,碘乙酰胺的烷基化半胱氨酸肽段数量最高,半胱氨酸烷基化和副反应不完整的肽段数量最少。与碘乙酰胺的烷基化条件被进一步优化。为了鉴定具有半胱氨酸的更多肽,使用硫丙基-琼脂糖6B树脂富集它们,并采用最佳条件进行还原和烷基化。富集产生的含半胱氨酸的肽多于没有富集的含半胱氨酸的肽的三倍以上。
更新日期:2017-11-21
中文翻译:
评估和优化还原和烷基化方法,以最大程度地利用基于MS的蛋白质组学鉴定多肽
质谱(MS)已成为分析蛋白质的越来越重要的技术。在流行的自下而上的基于MS的蛋白质组学中,还原和烷基化是促进肽鉴定的常规步骤。但是,可能会发生不完全反应和副反应,从而损害了实验结果。在这项工作中,我们系统地评估了常用试剂即二硫苏糖醇,2-巯基乙醇,三(2-羧乙基)膦或三(3-羟丙基)膦的还原步骤,以及碘乙酰胺,丙烯酰胺,N的烷基化-乙基马来酰亚胺或4-乙烯基吡啶。通过使用酵母全细胞裂解液中消化的肽,使用四种不同的还原剂,鉴定出的蛋白质和肽的数量非常相似。然而,四种烷基化试剂的结果与碘乙酰胺截然不同,碘乙酰胺的烷基化半胱氨酸肽段数量最高,半胱氨酸烷基化和副反应不完整的肽段数量最少。与碘乙酰胺的烷基化条件被进一步优化。为了鉴定具有半胱氨酸的更多肽,使用硫丙基-琼脂糖6B树脂富集它们,并采用最佳条件进行还原和烷基化。富集产生的含半胱氨酸的肽多于没有富集的含半胱氨酸的肽的三倍以上。


























 京公网安备 11010802027423号
京公网安备 11010802027423号