
Abstract
Christian Junge (1912–1996) is considered by many to be the founder of the modern discipline of atmospheric chemistry. In studies from the 1950s through the 1970s, Junge was able to link chemical measurements in a few scattered locations around the earth and integrate them with meteorology to develop the first global view of the basic chemical and physical processes that control the sources, transport, transformations, and fate of particles and gases in the atmosphere. In this paper we summarize and comment upon a number of Junge’s seminal research contributions to atmospheric chemistry, including his discovery of the stratospheric sulfate layer (known as the Junge layer), his recognition of the relationship between the variability of the concentrations of trace gases and their atmospheric lifetimes, his studies of aerosol size and number distributions, his development of the first quantitative model of tropospheric ozone, and other significant scientific investigations. We also discuss Junge’s professional life, his many international leadership positions and honors, as well as some memories and reflections on his many abilities that led to his outstanding contributions to the science of atmospheric chemistry.
Similar content being viewed by others
1 Introduction
Atmospheric chemistry plays a central role in global climate, weather, air quality, human health, and terrestrial and marine ecosystems. For example, the growing interest in climate, beginning in the mid-twentieth century, was accompanied by a great increase in research on the chemical properties of the atmosphere. The field attracted scientists from a broad range of disciplines, many of whom would later come to be regarded as pioneers in atmospheric chemistry. One of these was Christian Junge, a German meteorologist who had a unique impact on the development of atmospheric chemistry by integrating chemical studies with his knowledge of meteorology, a linkage that produced for the first time a global view of atmospheric chemistry. He excelled at taking chemical measurements made in a few scattered locations around the globe and integrating them to develop a global picture of the basic chemical and physical processes that control the sources, transport, reactions, and fate of particles and gases. Although this global picture of chemical cycles was tentative, often little more than a conceptual framework, it provided the foundation for the development of a global strategy for research on the chemistry of the atmosphere. This strategy focused on understanding the chemistry of the atmosphere to the degree that a predictive capability could be developed, enabling the determination of the impacts of societal decisions on the global atmosphere and Earth’s climate system. Because of his great impact in this emergent field of study, he is regarded by many as the founder of the science of atmospheric chemistry (see Fig. 1).

Christian Junge in Tenerife in 1968 (Photo and permission from Ruprecht Jaenicke)
Possibly many members of the current generation of the atmospheric chemistry and climate community may be unaware of the seminal role that Junge played in developing the discipline of atmospheric chemistry and its strategic goals. Now, near the 25th anniversary of Junge’s passing in 1996, we think it is appropriate to summarize and assess his contributions to the discipline and how they have contributed to our understanding of the atmosphere today. To this end, the review presented here was prepared largely by scientists who knew Junge and his research, including several who actively worked with Junge during his research years.
This paper first focuses on Christian Junge’s life. That is followed by discussions of ten seminal contributions that Junge made to the development of the modern discipline of atmospheric chemistry. These include the stratospheric sulfate (Junge) layer, aerosol number and mass size distributions, the linkage between the variability of trace gas concentrations to their atmospheric lifetimes, the first quantitative model of tropospheric ozone, the first precipitation chemistry network in North America, and his influential book Air Chemistry and Radioactivity published in 1963, among other topics.
These discussions are followed by a section on Junge’s leadership of international organizations and the advancement of atmospheric science, particularly atmospheric chemistry, as well as the many honors that he received. This is followed by a section on Memories and Reflections that focuses on Junge’s uniquely personal approach to scientific work, his leadership style, decision making, scientific humor, and how he encouraged and supported young scientists to develop their interests and research in atmospheric chemistry. Some material presented in this paper has been drawn from a shorter account of Junge’s life published in the German language (Jaenicke 2012).
2 Christian Junge’s life
Christian E. Junge was born in Elmshorn, Germany on July 2, 1912. Junge had an early interest in flying which later motivated him to study meteorology at the University of Graz, Austria. He continued his meteorology studies in Hamburg and Frankfurt from 1931 to 1935. He completed his Ph.D. at the University of Frankfurt (Junge 1935) under the supervision of Professor Franz Linke, studying atmospheric condensation nuclei. In Frankfurt, he spent two additional years working on condensation nuclei, and then joined the “Reichswetterdienst” (German Weather Service) at the Office for Instruments in Hamburg. Junge’s research career was heavily influenced by world politics and wars in the first half of the twentieth century. The path he followed did not always agree with his wishes. It was often detoured by contemporary social issues. For example, as a youth he had a strong interest in chemistry, but he selected meteorology as a career partly because he had seen many unemployed chemists looking for work during the Great Depression of the late 1920s and 1930s.
With the onset of World War Two, Junge’s work in the weather service led to his involvement with meteorology while serving in the Luftwaffe. Junge spent the entire war as a meteorologist on duty in many parts of the war theater, including Libya, Crete, Italy, and France. That duty helped shape his views on meteorology and the atmosphere. Later, he was held as a prisoner of war for two years, during which time he became severely ill. He eventually returned to the “Deutscher Wetterdienst” in Hamburg. His professional position there was that of an experienced weather forecaster and a trained atmospheric physicist with an interest in chemistry. That laid the groundwork for his later scientific career. In 1953 Junge returned to the University of Frankfurt for habilitation (i.e., a second dissertation and a permit to teach in universities) and he became “Privatdozent” – the highest university degree. These years lead to remarkable progress in atmospheric aerosols (Junge 1952, 1955).
Later in 1953, Junge joined the US Air Force Cambridge Research Laboratories (AFCRL) in Bedford, Massachusetts. While in the US, Junge conducted pioneering studies of many aspects of atmospheric chemistry including marine aerosol composition, the chemical composition of rainfall and the stratospheric aerosol layer. Eventually his research interests and those of AFCRL diverged and he left in 1961.
Back in Germany in 1962, Junge became a Professor of Meteorology at the University of Mainz. In that position, he had complete autonomy in his research. He was obligated to teach only six hours per week, and he could apply for research grants. Collaboration with Kurt Bullrich (1920–2010) and Hans-Walter Georgii (1924–2018) began early in Frankfurt, and when Junge returned to Mainz in 1962, they were among the first to start a “Collaborative Research Centre” in the German Research Foundation (DFG). He continued investigating trace gases (e.g., CO, H2, N2O) and aerosols (e.g., their size distribution and acidity), and developed a highly sensitive ion counter. His objective was to document the concentrations of those trace substances worldwide and determine their sources, budgets, and effects on the environment. A meteorologist with his imagination, experience and knowledge of atmospheric transport was ideally positioned to initiate such a broad field of investigations in those early years.
In 1968 Junge was appointed director at the Max Planck Institute (MPI) for Chemistry in Mainz, where he established the Department of Air Chemistry. At the MPI for Chemistry, Junge subsequently developed what was generally regarded as the leading international research program in atmospheric chemistry. Junge offered young scientists an outstanding environment to develop their careers. Young scientists from many different fields joined Junge’s group, which thereby grew rapidly in breadth and depth. Junge organized his program into four research sections: atmospheric aerosols, trace gases, reaction kinetics, and the “old atmosphere” (“alte Atmosphäre “, i.e., the long-term physicochemical evolution of Earth’s atmosphere). Figure 2 shows Junge’s group in Mainz in 1972. His work in Mainz significantly advanced our understanding of atmospheric physics and chemistry.

Christian Junge’s research group at the Max Planck Institute in 1972. Individuals numbered as follows: 1 Karl-Heinz Liebl; 2 Peter Warneck; 3 Peter Neuling; 4 Manfred Schidlowski; 5 Ulrich Schmidt; 6 Jürgen Hahn; 7 Frau Kuhning; 8 Wolfgang Seiler; 9 Karl Greese; 10 Renate Respondek (secretary); 11 Frau Schmidt (secretary); 12 Peter Winkler; 13 Werner Fiebiger; 14 Franz Slemr; 15 Reiner Beck; 16 Rudolph Eichmann; 17 B. Betz; 18 temporary visitor from India; 19 Grigorios Ketseridis; 20 Lothar Schütz; 21 Ruprecht Jaenicke (Photo and permission from Lothar Schütz)
Junge was a pioneer in climate issues. In 1971 he participated in a 3-week intensive working group that assessed the present and potential future climatic effects of human activities. The meeting included 30 of the world's leading atmospheric scientists from 14 countries. The resulting publication, “Inadvertent Climate Modification – Report of the Study of Man’s Impact on Climate” (SMIC 1971) played a major role in catalyzing concern and subsequent research on the human impacts on climate. The founding of the Max Planck Institute for Meteorology Hamburg in 1975 rests on Junge’s initiative. His influence was felt at many levels.
Junge’s aerosol research expanded into organic compounds, desert aerosols, and most particularly aerosols in the remote and largely undisturbed atmosphere. His motivating objective was to document the natural state of the atmosphere, which was becoming increasingly impacted by humans, even in remote regions. At that early date he was speculating about the influence of society on the evolution of the Earth’s atmosphere (Junge 1975a). Later, Paul Crutzen (1933–2021), the atmospheric chemist and 1995 Nobel Laureate and his successor as Director of the Air Chemistry Department at MPI, would further develop this idea into the concept of the “Anthropocene”.
A compilation of all Christian Junge’s scientific publications can be found at: https://www.mpic.de/5124255/junge-publications
Christian Junge retired in 1979, and he and his wife Ingeborg moved close to the residence of one of his two daughters at Lake Constance in southern Germany. There he followed his favorite leisure pursuits-history and anthropology. Christian Junge passed away on June 8, 1996, in Überlingen surrounded by his wife and his family. The life events, accomplishments, and key scientific contributions of Christian Junge are summarized in Table 1.
3 Seminal scientific contributions to atmospheric chemistry
3.1 The stratospheric Junge layer
Until the 1950s, research on stratospheric aerosols was largely motivated by an interest in the influx rate of extraterrestrial meteoritic debris (Opik 1956). Based on twilight measurements, Penndorf (1954) and Bigg (1956) had speculated about the vertical distribution of dust and the presence of a stratospheric dust layer. However, at that time much of atmospheric chemistry research was focused on the troposphere, especially on regions where the impact of pollution was most evident. Interest in the upper atmosphere increased rapidly in the 1950s largely because of two events. One was the launch of the Soviet Union satellite, Sputnik, in 1957, an event that triggered the space race and stimulated interest in the upper atmospheric environment. The other was the increasing concern about the fate and impact of radioactive debris produced in nuclear weapons tests that had increased dramatically during the 1950s and continued, though with greatly diminished frequency, until 1980.
In his position at the AFCRL, Junge’s initial focus on the stratosphere was motivated by concerns that micrometeorites might pose a threat to military satellites. Junge proposed an atmospheric aerosol research program. The proposal was accepted, and he was given abundant resources. The program began inauspiciously when an Air Force officer contacted Junge and offered to include his instruments on some planned stratospheric balloon flights without providing Junge with information about the flights or allowing him to participate in the planning. Weeks later, Junge was surprised to receive flight data and impactor samples collected from balloons that had been launched near Sioux Falls, South Dakota, carrying an Aitken particle counter and impactors. These data led to the first publication (Junge and Manson 1961) that presented convincing evidence of the presence of a stratospheric layer of particles. (Fig. 3).

Five vertical profiles of aerosol concentration measured in balloon flights near Sioux Falls, South Dakota, in 1959 and 1960 along with the height of the tropopause during the flights. The figure also shows profile measurements made by others in the troposphere (From Chagnon and Junge (1961), as shown in Junge (1963), Fig. 47, with permission from Elsevier)
The balloon studies were soon followed by a series of research flights on U-2 aircraft in 1960 (Junge and Manson 1961) using more advanced sampling techniques including improved impactors. The U-2 sampling programs enabled much greater temporal and spatial sampling capabilities and provided definitive measurements of an enhanced aerosol layer at about 20 km altitude (Junge et al. 1961a b). Because of his interest in chemistry, Junge also measured the chemical composition of particles collected in the profiles (Chagnon and Junge 1961) and found that \({\mathrm{SO}}_{4}^{2-}\) was the dominant chemical species in the stratospheric aerosol layer (Fig. 4).

Sulfur density as a function of latitude and altitude. The numbers refer to mass concentration in units of 10–15 g cm.−3 (From Junge and Manson (1961), Journal of Geophysical Research, permission not required by the American Geophysical Union)
The vertical profiles, coupled with an assessment of aerosol size and residence times, further suggested that the source of the particles in the layer was terrestrial, not extra-terrestrial. It is perhaps indicative of Junge’s mindset that the most comprehensive paper in this series (Junge et al. 1961a) was published in the Journal of Meteorology. Later work (Crutzen 1976) would show that, except for volcanic eruptions, the sulfur in the Junge layer was derived from carbonyl sulfide transported through the tropopause into the stratosphere where it is oxidized to sulfate. It should be noted that these measurements were made during the nuclear weapons testing moratorium in the late 1950s and during a period when there had been no major volcanic eruptions for decades, thus minimizing the impact of inputs from such events.
Because of his definitive work in these four papers, all published in 1961, the stratospheric aerosol layer carries Junge’s name. The aerosol in this layer continues to play a key role in the chemistry and dynamics of the stratospheric ozone layer and in climate research today (Brühl et al. 2012).
Junge played a unique role in characterizing the existence of the stratospheric aerosol layer. However, Junge played no role in the naming of the layer. The name “Junge layer” does not appear in the literature until the 1970s, the first occurrence apparently in Castleman et al. (1974). Hofmann and Rosen (1981) in a review that highlights the 20th anniversary of its discovery by Junge, emphasize the informal nature of the naming of the layer: “This ubiquitous 20 km feature has become known as the stratospheric aerosol layer, sulfate layer, or Junge layer.” Unfortunately, the details of Junge’s role in the discovery of the layer and the historic context of that work are not widely recognized today. A Wikipedia search on “Junge layer” is redirected to “Stratospheric sulfur aerosols” (Wikipedia 2022). In that article, the “Junge layer” is mentioned but Christian Junge is not and none of his pioneering work is cited.
3.2 Aerosol number and size distributions
At mid-century, the understanding of atmospheric aerosols was that air parcels contained a collection of apparently independent particle populations comprised of particles of different sizes and composition: Aitken particles, small ions, small-medium ions, large ions, Langevin ions, ultra-large ions, and haze particles (Israel and Schulz 1932). Junge had taken an interest in assessing the diverse aerosol size distribution data and in developing a coherent large-scale picture of these data. He attempted to rationalize these various data by using the same techniques that he had used in his Ph.D. studies in the analysis of atmospheric turbidity measurements (Linke 1942). At the time, the scientific literature was growing rapidly but much data consisted of discrete, geographically distributed measurements which were often difficult to interpret. An analogous situation existed with the interpretation of “line spectra” which were being obtained in many fields of research (e.g., atomic line spectra). This approach seemed reasonable because at the time particle physics research was yielding a large array of apparently unrelated particles as well.
Junge’s work as a weather forecaster during the war had required him to deal with sparse and diverse weather data. It was difficult to prepare forecasts because meteorological data were kept secret by the combatant nations. However, the safe return of aircraft depended on good weather forecasts. Because Junge had only sparse pressure data to work with, he became adept at drawing synoptic pressure (isopleth line) maps, in which he bridged data gaps using his broad meteorological knowledge and insights. Junge applied this same procedure to the analysis of aerosol data. He wrote, “For simplicity, the line spectra [discrete bin-size measurements of particles] were converted to continuous distributions” (Junge 1952). In this way, he proposed an equation that described a continuous aerosol number size distribution that yields concentration density as a function of radius (Fig. 5). He wrote,”… the most striking feature of these ‘continental’ size distributions is the regular decrease in a concentration above 0.1 µm radius, which can be approximated by dN/d(log r) = cr−β, where β is about 3 and c is a constant.” If β is 3, then the corresponding mass or volume distribution is constant for equal logarithmic intervals of radius, a relationship that was generally valid up to the maximum radius considered at that time, 20 µm.

Its simplicity was so convincing that colleagues accepted the proposed equation as a “law”. Probably this view was popularized by a remark made by Heinrich Friedrich Siedentopf (1906–1963) at a presentation by Junge. Siedentopf, a German astronomer and physicist, commented, “An r−3 law of radii distribution is not only present in atmospheric dust, but also in interstellar material.” However, there is a flaw in this “law”. If \(\beta =3\), c has the dimension 1, because dN/dlogr has units of cm−3 (dN/dlogr is a concentration density). The scientific community was so impressed with this simple relationship that they failed to notice the problem with c. If β is not 3 - Junge had written “…is about 3…”- then c has a dimension unequal to 1. Later, Jaenicke and Davies (1976) added a more detailed mathematical formalism. The latter might be more correct, but Junge’s “law” had the advantage of simplicity and it continued to be used for some time in the aerosol community.
This simple picture of aerosol size distributions dominated the aerosol research community for many years. However, as more measurements were made using greater size and composition resolution, a much more complex and dynamic picture evolved. Co-author Jaenicke recalls a discussion with Kenneth Whitby (1925–1983) at the University of Minnesota in 1971. Whitby’s laboratory was focused on making state-of-the-art aerosol measurements. Whitby questioned the validity of Junge’s double logarithmic straight lines. In a seminal work, Willeke and Whitby (1975) showed that aerosol size distributions are typically multimodal, reflecting inputs from a variety of sources, both primary and secondary, and also removal by processes that are highly size-dependent. It is essential to understand these processes if one is to model aerosol size distributions and to account for changes in chemical as well as physical properties (e.g., Whitby and McMurry 1997). Nonetheless, Willeke and Whitby show (see Fig. 6) that the gross features of their multimodal distributions follow in a general way Junge’s power law over three orders of magnitude in size on a log–log plot.

The grand average of the number aerosol size distribution from October 1971 measurements at Denver’s City Maintenance Yard (solid line) compared with a Junge power-law distribution (dashed line) calculated using the constants of Clark and Whitby (1967) (From Fig. 1 of Willeke and Whitby (1975), with permission from Taylor and Francis)
3.3 Variability and lifetime of tropospheric trace gases
In 1974, Junge published a seminal paper that linked the time and space variations of trace gases in the atmosphere in an effort to characterize their chemical behavior on a global scale (Junge 1974). He defined the expressions “lifetime” and “residence time” of gases in the tropospheric reservoir. The “lifetime” refers to the removal rate by chemical destruction, including deposition to the surface, and “residence time” to transport into other reservoirs, e.g., between the troposphere and the stratosphere. Junge then generalized the term “residence time” to include both aspects. For chemically reactive species transport to the stratosphere is usually of minor importance and many authors have limited the discussion to the lifetime, τ, which is also adopted here.
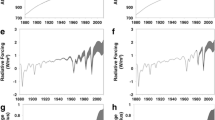
Junge’s effort to relate the variability of trace gases to their lifetime was motivated by his observation that concentration fluctuations decrease with increasing τ (Junge 1963, page 1). The fluctuations were expressed by the relative standard deviation σ′ (i.e., the regular standard deviation σ divided by the mean) of the measured concentration of a compound, which was found to be inversely proportional to τ (Fig. 7). From his measurement data, presented in prior publications, Junge derived the following empirical relationship between the variability of compound X and τ (in years):

The variability-lifetime concept principally applies to all trace gases with uniform source and sink functions, assuming first-order removal processes (Gibbs and Slinn 1973). In practice, however, sources and sinks can be quite inhomogeneously distributed. Hamrud (1983) applied a global, two-dimensional (zonally averaged) numerical model of the troposphere and stratosphere, testing different source distributions. He concluded that if the locations of sources and sinks are unknown, the lifetime of a trace gas derived from its variability can be uncertain by at least an order of magnitude. But if the locations are well-defined, the uncertainty is greatly reduced to about a factor of two (Hamrud 1983). Slinn (1988) showed that for a sampling time between one month and one year, Junge’s empirical and Hamrud’s numerical calculations are generally consistent.
The relationship (1) has been applied in many studies. For example, it was used to estimate the lifetime of carbonyl sulfide (COS), which is chemically converted to \({\mathrm{SO}}_{4}^{2-}\) in the stratosphere where it contributes to the Junge layer (Junge et al. 1961a; Crutzen 1976). The lifetime of COS in the atmosphere by chemical breakdown was estimated to be rather long, ~ 35 years. However, measurements of temporal variability at Amsterdam Island in the late 1980s indicated a τ of about 1.4 years, close to the more recently derived τ of ~ 2 years (Mihalopoulos et al. 1991). The τ of COS is now known to be governed through uptake by vegetation (Brühl et al. 2012). Jobson et al. (1999) advanced the concept to study the regional lifetime of non-methane hydrocarbons through the reaction with OH radicals, based on measurements in urban and remote locations. For these relatively short-lived compounds, with a typical τ of days to months, they followed the suggestion by Jaenicke (1982) that the Junge relationship can be refined by an exponential decay function, replacing σ′ by the regular standard deviation of the logarithm of the concentration, σ(ln X), through the expression:
A is a fitting parameter and b relates to the regional sink terms, influenced by upwind mixing processes. The expressions (1) and (2) become equivalent for long-lived species, i.e., \(\sigma \left(\mathrm{ln}X\right)\approx {\upsigma }^{\mathrm{^{\prime}}}\left(X\right).\) The parameter b is typically about 0.5 in the background troposphere and can vary between 0 in urban sites, where the variability is determined by local emissions, to about 0.5 or even higher in very remote areas, where the variability solely depends on chemical loss (Ehhalt et al. 1998; Jobson et al. 1999). Williams et al. (2000) took Jobson’s approach a step further to help identify unknown hydrocarbon compounds measured in the atmosphere by mass spectrometry. In this way, it is possible to detect outliers and evaluate the quality of datasets including contamination issues, to estimate OH concentrations, and to distinguish between the chemical breakdown products by OH and those by halogen radicals. They also suggested that the parameter A is a measure of the range of elapsed times between the emission of a compound from a source and its detection at the receptor site, i.e., the chemical “age” of the transporting air masses.
Junge’s variability-lifetime concept has proven to be valid on various scales, through different formulations for many compounds and in many settings, mostly for gases but also for aerosols (Williams et al. 2002). Junge introduced it because of his interest in global atmospheric chemistry. Because of the limited datasets available at the time, he and others applied the concept to the analysis of campaign data. Warneck (2000, p.162) concluded that helium appeared as an outlier in Fig. 7 in the original work of Junge (1974) because of limitations in the measurement techniques. When measurements of trace gases are associated with spurious variability (noise) or if atmospheric concentrations are increasing rapidly (i.e., not being in steady-state) their lifetime can be underestimated, which may have happened with N2O and CFCs in the 1970s and 1980s. Therefore, the method needs to be applied with caution. Nevertheless, in the past half-century, the originality of Junge’s concept has been widely recognized. His concept of lifetime has become an integral part of atmospheric science textbooks. It has been applied, with adjustments, to environmental studies and has even been adopted in other disciplines.
3.4 Chemical composition and processing of aerosol particles in the remote troposphere
Junge’s early studies of the activation of condensation nuclei into cloud droplets based on size and chemical composition (e.g., Junge 1951) led to investigations of factors that modulate the size-resolved composition of tropospheric aerosols. Initially, two aerosol size fractions (0.08 - 0.8 and 0.8 - 8 µm radius, nominally “large” and “giant” nuclei, respectively) were sampled near Frankfurt, Germany, on the rural New England coast, and in the vicinity of Boston; these were analyzed for major ionic constituents (Junge 1953, 1954). In addition, to evaluate the anthropogenic influence on aerosol composition in polluted regions and to investigate differences in marine versus continental aerosol composition, he characterized the same aerosol size fractions in near-surface marine aerosol collected at Homestead, Florida, and Hilo, Hawaii. In Hawaii, the atmosphere was also sampled over a vertical gradient from sea level to ~ 2900 m. To investigate potential multiphase chemical interactions, particulate measurements in Florida and Hawaii were augmented with paired measurements of alkaline-reactive trace gases (oxidized and reduced N, S, and Cl) (Junge 1956, 1957). These were the first simultaneous multiphase observations of reactive species in the Earth’s atmosphere. The Hawaiian measurements were made in the context of a broad multidisciplinary study, Project Shower, which was “conceived as a means for obtaining measurements which could acquire extended meaning as they were studied in relationship to each other.” “Measurements such as these were for the most part already available for separate conditions, but no single working group in cloud physics had been formed and equipped to gather such information at one place and time” (Editor, Tellus, 1957).
Results were interpreted in a meteorological context based on source region (marine versus continental), air mass history (transport path, proximity to pollution sources, precipitation history), altitude (marine boundary layer – MBL-versus free troposphere-FT), and variability in constituent ratios. Junge regarded many of his interpretations of these novel data to be preliminary and some have not withstood the test of time with the emergence of higher-resolution data and our increased understanding of processes. However, in many respects, his multifaceted approach to experimental design and data analysis laid the groundwork for future studies. Junge’s effective interpretation of results based on rudimentary back trajectories inferred from weather maps and wind velocities pointed to the need for the development of refined atmospheric-transport models (e.g., Stohl et al. 1998). Based on ratios of constituents relative to those in seawater, Junge correctly inferred that virtually all particulate \({\mathrm{Cl}}^{-},\; {\mathrm{Na}}^{+},\;\mathrm{ and }\;{\mathrm{SO}}_{4}^{2-}\) associated with “giant” particles in marine air originated from seawater. Analogous approaches for differentiating sea-salt and non-sea-salt components of marine aerosol and precipitation have since been widely employed by the research community (e.g., Keene et al. 1986). The low \({\mathrm{Cl}}^{-}\) to NH4+ ratios associated with particles sampled above the inversion layer at Hawaii suggested that FT aerosol in marine regions did not necessarily originate from seawater. Nitrate in marine-influenced air was associated almost entirely with “giant” particles and concentrations were highest in polluted coastal air, which suggests rapid chemical interactions between nitrogen oxide precursors and marine aerosol. Finally, Junge’s strategy of using measurements from remote regions as a benchmark to evaluate anthropogenic influences on atmospheric composition in polluted regions has been widely employed in subsequent studies (e.g., Galloway et al. 1984).
An important insight gained from this early work was the recognition that the absolute and relative concentrations of size-resolved particulate constituents and the associated gas-aerosol phase partitioning vary significantly over space, time, and air mass history. This led to the prescient conclusion that “… a real step forward in the understanding of the basic processes in air chemistry can be gained only if aerosols and gases are measured simultaneously but separately, and if the aerosols, in turn, are separated according to size…” (italicized for emphasis by Junge 1956). This basic approach to process-level investigations of the chemically coupled multiphase system served as the model for future studies and a key foundation for progress in the field.
In two seminal reviews, Junge integrated the results of his work with those of others into a broader, global-scale perspective (Junge 1958, 1959). He correctly deduced that bursting bubbles from breaking oceanic wind waves are the major global source of aerosol mass and that this mass is dominated by inorganic sea salt that is associated primarily with particles greater than 1.0 µm radius. However, the inferred numbers of “sea-spray” particles were one to two orders of magnitude less than the corresponding number concentrations of condensation nuclei. This observation led Junge to initially speculate that most Aitken particles over the remote ocean originate from continental sources. Subsequent papers elaborate on the hypothesis that there is a fairly uniform “background aerosol” population over the remote ocean that originated from two processes: Transport from distant continental sources and the nucleation of new particles from precursor gases within the MBL (Junge et al. 1969; Junge 1972; Peterson and Junge 1971). Recent studies now support the hypothesis that number size distributions and corresponding environmental implications of aerosol in the remote MBL are modulated primarily by the physicochemical evolution of organic-rich particles produced via bubble bursting and of particles entrained into the MBL from the FT (e.g., Sanchez et al. 2018). Junge (1972) described the atmospheric processing of marine aerosol as due to: coagulation; cloud processing including the aqueous-phase oxidation of SO2 to H2SO4; the reaction of NO2 with marine aerosol; the condensation of NH3, H2SO4, and organics from the gas phase; and wet and dry deposition.
Junge et al. investigated the composition of particulate organic matter (OM) sampled in bulk over Northern Europe and in relatively clean marine air over the North Atlantic Ocean (Ketseridis et al. 1976). Concentrations of total organic carbon (TOC) and ether-extractable organic carbon (EEOC) in marine air uninfluenced by Saharan dust were lower than those over Europe. In contrast, the relative variability of individual components of EEOC including organic acids and phenols, organic bases, aliphatic hydrocarbons, aromatic hydrocarbons, and neutral compounds was similar in both polluted and marine air. The authors posed two possible explanations for these results. (1) Most particulate OM is produced in polluted regions and is transported over the oceans diluted but essentially chemically unaltered over the ocean. (2) Alternatively, particulate OM is produced in marine regions, but its chemical composition is similar to that produced in polluted regions.
In follow-up investigations, a suite of alkanes and aromatics were measured simultaneously in the vapor phase and in bulk aerosol together with particulate TOC, EEOC, and components of EEOC on the west coast of Ireland and at Cape Grim, Tasmania. (Eichmann et al. 1979, 1980). These studies produced the first paired multiphase observations of atmospheric OM. Based on measurements at Cape Grim, Tasmania, and the estimated atmospheric lifetimes of gas- and particulate-phase species (4 to 5 days) coupled with the long upwind trajectories over the ocean at Cape Grim, Junge et al. concluded that both gaseous and particulate alkanes are of marine origin. The similarity between alkanes, EEOC, and EEOC components in onshore flow on the west coast of Ireland and at Cape Grim implied that OM at the Irish site also originates from the ocean. Out-of-sector (non-oceanic) flow at Ireland was associated with higher concentrations of alkanes and aromatics suggesting continental sources. Eichmann et al. conclude that the incorporation of reaction products from the oxidation of marine-derived precursor gases is the primary source of particulate OM mass in marine air. Subsequent work by other groups confirms the importance of this pathway (e.g., Saltzman et al. 1985). Considering their findings, Eichmann et al. (1980) characterized the uptake and processing of OM associated with marine aerosol as “a wide-open question.” Our current understanding of these pathways is still incomplete and remains the focus of ongoing research.
3.5 First precipitation chemistry network in North America
In the mid-fifties, Junge et al. established the first North American deposition sampling network that focused on characterizing the chemical composition of precipitation (Junge and Gustafson 1957; Junge and Werby 1958). As previously noted, Junge had a unique ability to analyze relatively simple data sets and extract insightful conclusions. An excellent example is his analysis of the concentration of soluble species in precipitation collected in a one-year study (July 1955 through July 1956) in a broad network of sixty-two stations distributed across the entire United States (Junge and Gustafson 1957; Junge and Werby 1958; Junge 1958). Maps depicting contour plots of aqueous concentrations of sea salt (as its major constituent ions \({\mathrm{Na}}^{+}\) and \({\mathrm{Cl}}^{-}\)), sulfate (\({\mathrm{SO}}_{4}^{2-}),\) nitrate (NO3−), ammonium (NH4+), potassium (K+), and calcium (Ca2+) revealed regional patterns that could be linked to sources, including pollution and the occurrence of “acid rain”, although the authors never used that term. The seminal paper by Junge and Werby (1958) correctly identified deflation of surface soils as the primary source of Ca2+ and presumably K+ in continental air; we now know that biomass burning is also a major source of K. Junge appears to be the first to recognize that much of atmospheric SO42− over large areas of the United States is associated with sea salt, which he correctly surmised originates from sea spray and bubble bursting. He also plotted contour maps of “excess sulfate,” by correcting for sea salt via normalizing based on \({\mathrm{Na}}^{+}\). These maps clearly show the impact of anthropogenic activities in the Ohio River Valley and other heavily industrialized areas (see Sect. 3.8).
The beautiful maps of \({\mathrm{Na}}^{+}\) and \({\mathrm{Cl}}^{-}\) concentrations showed a large gradient with the highest near the coasts decreasing to generally low concentrations inland, but occasional hot spots in places such as the salt flats of Texas and the arid west. The measurements also revealed a \({\mathrm{Cl}}^{-}\) deficit relative to the molar ratio of Cl/Na near unity (~ 0.86) in sea salt. Junge attributed this \({\mathrm{Cl}}^{-}\) deficit to extra \({\mathrm{Na}}^{+}\) from wind-blown mineral dust. Recent work shows that acid-displacement, free-radical, and other multiphase chemical transformations involving particulate \({\mathrm{Cl}}^{-}\) in the atmosphere produce inorganic chlorinated products including HCl, \({\mathrm{ClNO}}_{2},\;\mathrm{ BrCl},\;\mathrm{ and }\;{\mathrm{Cl}}_{2}\) that volatilize to the gas phase (e.g., Long et al. 2014). The dry-deposition fluxes of particulate and volatile Cl species differ significantly and also some of the volatile species have relatively low solubilities. Consequently, the total amounts of particulate and volatile Cl scavenged and deposited by precipitation may diverge from the sea-salt ratio with \({\mathrm{Na}}^{+}\) independent of contributions from non-sea-salt sources. Therefore, ratios of \({\mathrm{Cl}}^{-}\) to \({\mathrm{Na}}^{+}\) in precipitation relative to those in seawater cannot be reliably interpreted in terms of sources (Keene et al. 2015). Evidence based on ensembles of constituent ratios and associated transport modeling supports Junge’s hypothesis that the deflation of surface soils contributes to \({\mathrm{Na}}^{+}\) and \({\mathrm{Cl}}^{-}\) in precipitation at inland continental locations (Jordan et al. 2015). The consistency of Junge’s results with modern measurements affirms the quality of his analytical methods for \({\mathrm{Na}}^{+}\) and \({\mathrm{Cl}}^{-}\). It also demonstrates the intellectual integrity of the author because one might be tempted to assume consistent composition of sea salt. Junge was not the first to examine deposition data and to speculate on sources and impacts. The best early examples of such work were the rainfall chemistry studies carried out in the Nordic countries (e.g., Odén 1968). However, these networks were limited in their areal coverage and could not be readily generalized to global scales.
3.6 First quantitative model of tropospheric ozone
Prior to the 1960s, there was no comprehensive conceptual framework for understanding ozone in the troposphere. Our knowledge consisted of a collection of disparate facts. That changed in the 1960s when Junge published the first attempt at balancing the global ozone budget (Junge 1962a; see also Junge and Czeplak 1968). These papers are limited in that they failed to identify the role of photochemistry as the dominant source and sink of tropospheric ozone. The importance of UV photolysis (Jones and Wayne 1970), and a seminal paper by Levy (1971), who identified the central role of the OH radical, were followed by the remarkable insights of Crutzen (1973) about the chemistry of ozone in the troposphere. Despite this omission, an examination of Junge’s publications with the benefit of 60 years hindsight reveals a masterly work that established a systematic framework for future tropospheric ozone research.
To appreciate the state of the science in the early 1960s and the magnitude of Junge’s contributions, consider what was known at that time: Ozone mixing ratios in the stratosphere are many times greater than in the troposphere and stratospheric intrusions can transport ozone-rich air into the troposphere. There are regular annual cycles in both stratospheric and tropospheric ozone. Ozone is destroyed at the Earth’s surface, a process that was then incorrectly assumed to fully explain its vertical profile in the troposphere (Regener 1957). Representative background concentrations could only be obtained by either sampling at the surface at mid-day and early afternoon when the atmosphere is usually well mixed or by sampling from exposed, elevated locations (Ehmert 1959).
Junge judiciously picked the most reliable of these observations away from sources of local pollution and transformed this field in his usual way by developing a conceptual framework that yielded a coherent picture (Junge 1962a; Junge and Czeplak 1968). Junge (1962a) represented these observations and processes in a simple mathematical model that included mass balance, the seasonal cycles of stratospheric ozone, stratospheric-tropospheric exchange, and ozone destruction near the Earth’s surface. Junge developed an analytical solution and found the best fit of the model to the facts. The key variable was the delay between the peak in the stratospheric injection of ozone into the troposphere and the peak in tropospheric ozone content. His best estimate was consistent with an ozone residence time of 3.3 months. This model provided an essential framework for ozone research. Along with the mathematical model, Junge (1962a) clarified three key concepts: hemispheric tropospheric ozone is well-mixed on time scales of months; the two hemispheres are essentially decoupled with respect to tropospheric ozone with opposite phases in their annual cycles; and tropospheric ozone has a quantifiable residence time or lifetime. These three concepts are so basic that today we often fail to recognize how innovative they were.
We now know that the lifetime of ozone is longer in the FT than in the planetary boundary layer (PBL) and therefore concentrations are less variable, validating Junge’s choice of data for quantifying the global budget. At the surface, the diel cycle of ozone concentrations is driven to a large extent by vertical mixing, a process correctly identified by Junge as a cause of the local ozone maximum at noon or afternoon, following on the earlier work of Auer (1939), Götz and Volz (1951), Teichert (1955) and Ehmert (1959).
Junge also addressed questions about the annual cycle. Many high-altitude clean-air stations show a spring maximum in ozone. Junge’s paper posits downward transport from the stratosphere as the cause of this cycle. The lifting of the tropopause with the arrival of spring is a contributing factor. Total column ozone (~ 90% of Earth’s ozone is in the stratosphere) shows a maximum in late winter or early spring when the tropopause is lowest. Ozone in the lower free troposphere reaches a maximum a few months later.
Junge verified these conclusions by noting a similar seasonal pattern in surface concentrations of radioactivity. Beryllium-7, a radionuclide (half-life of 53.3 days) is naturally produced by cosmic-ray induced spallation of N at high altitudes mainly in the stratosphere. Subsequently 7Be becomes attached to aerosol particles that are later transported down into the troposphere. Thus, 7Be can serve as a good tracer of transport from the stratosphere. Above-ground nuclear weapons testing led to mushroom clouds that carried radioactive debris, including long-lived fission products such as 90Sr, into the stratosphere where they became well-mixed. These long-lived fission products and 7Be are transported to the troposphere by gravitational settling followed by stratospheric-tropospheric exchange events. Measurements of their tropospheric concentrations and deposition provided Junge with independent evidence of the timing of the annual maximum of this stratospheric-tropospheric exchange and insight into the ozone budget.
In this context, Junge explored large-scale global exchange and mixing in the troposphere in three papers (Junge 1962b; Junge and Czeplak 1968; and Czeplak and Junge 1974) using CO2, tritiated methane and ozone as tracers. A pole-to-pole time-dependent mathematical line model with functions describing both horizontal diffusion and source terms as well as a derived two-box model both showed a good approximation to the data and allowed them to estimate an interhemispheric exchange time of about 400 days (Czeplak and Junge 1974). This explained why short-lived trace constituents, released in one hemisphere, are unable to effectively penetrate into the other hemisphere. The meteorological equator (ITCZ) forms an effective barrier for those substances, e.g., short-lived fission products. Junge characterized the essential time scales of large-scale tropospheric mixing within a hemisphere to be of the order of one month and between the hemispheres to be roughly one year.
In his 1962 paper, Junge called for the development of a meridional surface ozone monitoring network because “…tropospheric ozone may be a very powerful tool for studying the exchange mechanism between stratosphere and troposphere” and provide “better and useful information on the interesting and important subject of the blocking action of the equatorial circulation for large scale exchange between the hemispheres.” Perhaps the important concept that Junge expressed for that time was that for many areas of investigation, atmospheric composition needed long-term, high-quality observations. Today there are many national, multinational, and global monitoring networks including the World Meteorological Organization Global Atmospheric Watch Network (Schultz et al. 2015). To some extent, Junge’s call for a global ozone network has been achieved (Schultz et al. 2017). However, the recent analysis of data from one collection of surface ozone networks (Sofen et al. 2016) shows that at that time they effectively covered only 25% of the Earth’s surface (a good coverage for a surface network) but failed to provide representative ozone monitoring over the remaining 75%.
Many aspects of the tropospheric ozone budget have been clarified since 1962 (e.g., Archibald et al. 2020). The average lifetime of tropospheric ozone is much less than three months. Junge had concluded that smog photochemistry was limited to urban areas like Los Angeles, but we now know that ozone is produced wherever NO is present and the net O3 production (gas-phase O3 production minus gas-phase O3 loss) becomes positive at low NO mixing ratios – a few to a few tens of ppt depending on other species present. Above this compensation point O3 production predominates through reactions of NO with RO2 and HO2 (Fishman and Crutzen 1978) and net ozone production ensues. In remote locations especially parts of the marine boundary layer where NO concentrations are very low, photochemical reactions (notably those producing OH radicals) are the dominant loss process – they can destroy ozone faster than dry deposition to the ocean surface. In the second half of the 20th Century, anthropogenic NOx emissions from fossil fuel combustion and the use of fertilizers have upset the balance and disrupted the global ozone budget leading to widespread ozone pollution. In recent decades, there has been an encouraging decline in NOx emissions in North America and Europe, bringing a glimmer of hope for reducing smog and climate impacts (Krotkov et al. 2016). Deposition velocities over the oceans are slower than over land but halogens can play a role in ozone destruction in the marine boundary layer. Dry deposition over land is now known to be a richly complex process involving plant growth and transpiration and can have a profound impact on carbon sequestration. Starting in the 1970s, a debate raged over “meteorologists’ ozone” i.e., ozone essentially inert in the troposphere vs. “chemists’ ozone” with full tropospheric chemistry. But progress in atmospheric science has shown us that one must understand physics, chemistry, and their meteorological context (Lelieveld and Dentener 2000). Many trace gases important in ozone chemistry (e.g., H2O, NOx, CO, CH4, and other VOCs) have major sources in vegetation and other life forms. Thus, their atmospheric concentrations depend on the state of the vegetation and the health of the ecosystem. Therefore, studies of atmospheric composition are increasingly designed to also account for interdependencies with the biosphere.
3.7 Transport of African dust to the western hemisphere
Windborne mineral dust plays an important role in climate by modulating solar radiation and cloud microphysical properties which, in turn, affect radiation and the hydrological cycle (Ghan et al. 2012). Also, dust carries elements such as iron and phosphorus that serve as essential nutrients when deposited to marine and terrestrial ecosystems (Duce 1986; Jickells and Moore 2015). Today, there is great interest in North African dust because the largest and most persistently active dust sources are found there, supplying over half of total global emissions (Huneeus et al. 2011). Moreover, North Africa is expected to be greatly impacted by climate change, which would affect dust emission rates and transport and, in turn by feedback processes, climate.
Early researchers, including Darwin (1846), had commented on the great quantities of dust transported across the west coast of North Africa to coastal waters. African dust is known to have been transported over southern Europe for at least 2000 years based on glacial cores from the Alps (Clifford et al. 2019). Earlier, Hellmann and Meinardus (1901) had reported a period of dustfalls during spring 1901 extending from Northern Africa to the Baltic coast. However, there was no awareness of the possible impact that this dust might have on larger spatial scales. Junge was the first to report the observation of African dust in the Western Hemisphere (Junge 1956) during a study of sulfur and nitrogen species in gases and aerosols in South Florida noted above in Sect. 3.4, He had carried out studies at a coastal site near Round Hill, MA (ca., 41.5°N, 71.9°W) in 1953 and reported on a wide range of nitrogen, sulfur, and sea-salt species in aerosol (Junge 1954). Concerns about pollution impacts in the northeast led him to plan a study in Florida where anthropogenic impacts would be minimal. In July and August 1954, he made measurements in the Trade Winds at a coastal site near Homestead, Florida (25.5°N, 80.5°W), an agricultural region 40 km south of Miami (Junge 1956).
Junge’s paper focuses almost exclusively on nitrogen and sulfur species as discussed in Sect. 3.4. However, in one paragraph, buried deep in the paper, he mentions the presence of dust in his samples. He states, “It might be mentioned here as a matter of curiosity that during the last three days, beginning with 31 July, the aerosol samples of both sizes [“fine” and “coarse” size classes] contained considerable, though decreasing [over the following days] amounts of reddish-yellow dust.” This indication of dust is shown in Fig. 8 (Junge 1956).

The first identification of African dust in the Western Hemisphere, symbolically identifing a dust event on 31 July 1954. The letters “S” and “L” indicate whether the sample was collected under sea-breeze or land-breeze conditions. The dust plot scale has no units. (This figure is a copy of a portion of Fig. 1 from Junge (1956) in Tellus. Permission not required by Tellus)
Turning to synoptic weather maps, he noted that “Very steady easterly (trade) winds prevailed at all levels above the Atlantic Ocean during this period. It is very probable that this dust was carried across the Atlantic from West Africa, where several dust storms had occurred during the first half of July.” Recently, Prospero et al. (2021) performed daily back trajectory calculations (NOAA HYSPLIT, Stein et al. 2015) for Junge’s experimental period. These showed that only on one day, July 31, did back trajectories trace back directly to coastal North Africa, over a distance of 7000 km. They subsequently penetrated deeply into Mauritania and southern Algeria, thus validating the general conclusions made by Junge 70 years earlier.
Junge does not present any measurements of dust properties. He bases his conclusion solely on the size distribution of the dust (“fine” and “coarse”) and the color. Junge was well primed for his conclusions. During World War Two, as previously noted, he served as a meteorologist with the Luftwaffe stationed in Libya (Jaenicke 2012) (Sect. 2). There he would have become familiar with dust storms and the distinctive color of the dust. In his role as a weather forecaster, he would have been concerned about dust storms, which would have made flight operations hazardous. Based on this background, Junge stimulated measurements of the number and mass size distribution of desert dust and soils in Libya (Schütz and Jaenicke 1974).
Commenting from his experience as a meteorologist, he notes a problem with his conclusion about transport from Africa, “…, it must be assumed that the dust was carried all the way across the Atlantic Ocean below the inversion layer. It is surprising that it was not washed out by the trade-wind showers.” Later studies (e.g., Delany et al. 1967; Prospero and Lamb 2003) would show that African dust is carried to the Caribbean Basin on an annual cycle with a maximum in boreal summer. Transport over the Atlantic occurs primarily in the Saharan Air Layer (SAL), a hot, dry layer that lies above the marine boundary layer (Prospero and Carlson 1972; see also Prospero et al. 2021). The SAL inversion limits precipitation removal except in tropical disturbances. Even so, precipitation removal is the major removal mechanism over the tropical Atlantic and it presents a challenge to our understanding of long-range transport and the modeling of the entire dust cycle (Zhao et al. 2022).
Junge’s statement is the first in the scientific literature to identify African dust in marine aerosol samples collected in the western hemisphere and to credibly speculate on the possibility of transport from specific sources – dust storms – in a specific region. This experience may have primed him to be alert to the possibilities of long-range transport in his later studies and may have led him to think more globally in his research. Given the brief nature of the dust event, one wonders if a scientist with narrower interests would have focused much attention on it especially considering the absence of quantitative measurements. It is a credit to Junge’s broad-ranging interests that he recognized the significance of the dust in his samples. In later years Junge continued to have an interest in African dust. He noted, for example, that the size distribution of dust in the Caribbean (Delany et al. 1967) matched that from other remote locations and that it followed the power law shown in Sect. 3.2 (Junge 1955). Junge would continue to study African dust at an observatory on Tenerife and on Atlantic cruises of the German research vessel “Meteor” (Junge 1972).
3.8 Anthropogenic-driven changes in atmospheric composition and their impacts
A central focus of the atmospheric chemistry community today is to characterize and assess the impact of anthropogenic emissions on the composition of the atmosphere and the consequences for human and ecosystem health, weather, and climate. Junge’s early work played a major role in characterizing these impacts (e.g., see Sect. 2 for his involvement with the SMIC (1971) report on human impacts on climate). He showed that meaningful budgets can be created even for short-lived species. These could serve as the framework for understanding the magnitude and consequences of anthropogenically-driven changes in the composition and properties of the atmosphere.
In Junge’s 1963 book he points out the great difference between background concentrations of trace species and the values obtained in cities, where human impacts are maximal. In remote areas, concentrations of particulate matter (5–50 µg m–3) are low relative to urban areas with 100 s or even 1000 s of µg m–3. He further notes that the composition of aerosols in cities is dominated by combustion products including \({\mathrm{SO}}_{4}^{2-},\) NO3−, and OM. SO2 was a particularly important component of the polluted atmosphere in the mid-20th Century due to the widespread combustion of high-sulfur fossil fuels. Junge pointed out that this gas is rapidly converted to \({\mathrm{SO}}_{4}^{2-}\) that we now know contributes to the aerosol loading with consequences for cloud cover, precipitation, and radiative balance. He noted that iron concentrations were similar in urban and non-urban areas but that there was a great excess of lead in cities. This was attributable to tetraethyl lead, an anti-knock compound then used in gasoline. His early text also discussed the problem of eye-burning “oxidants” including ozone and how they are produced by the action of sunlight on NO2 in the presence of hydrocarbons, although the details of photochemical reactions were mostly unknown at the time, as described in Sect. 3.6.
Junge used data from the US precipitation network described in Sect. 3.5 to show impacts from human sources. The plot of excess-\({\mathrm{SO}}_{4}^{2-}\)(i.e., the concentration of \({\mathrm{SO}}_{4}^{2-}\) after subtracting the sea-salt \({\mathrm{SO}}_{4}^{2-}\) contribution based on the \({\mathrm{Na}}^{+}\) concentration) shown in Junge and Werby (1958) (see Fig. 9) is especially enlightening. It is immediately apparent that excess \({\mathrm{SO}}_{4}^{2-}\) is linked to continental sources, shown most clearly by the strong gradient along the east coast. One can also see clusters of high concentrations of excess -\({\mathrm{SO}}_{4}^{2-}\) in the northern states around the Great Lakes and the Midwest in the Ohio River Valley. There is a second grouping of high values in the Southwest, especially along the border with Mexico. Junge rightfully attributes these effects to emission from industrial activities except for the high values along the border with Mexico, which he attributes to soil inputs but which we now know are most likely linked to smelters, many in Mexico, and power plants. He discusses the various species and their ratios and reaches a wide-ranging series of conclusions that are speculative but insightful.

Junge ends with statements that illustrate his global view and his boldness in addressing the issues; “We think that enough data are now available to discuss the sulfur cycle on a global basis.” He concludes that “about 30 percent of this additional \({\mathrm{SO}}_{4}^{2-}\) on a global scale is due to human activities.” Junge correctly surmised that human sources made a substantial contribution to the global sulfur budget and precipitation acidity – they still do. Furthermore, he stated that the combustion of coal played a dominant role. Finally, he addressed climate impact, noting that particulate \({\mathrm{SO}}_{4}^{2-}\) plays an important role as condensation nuclei in the atmosphere and, consequently, pollution-derived particles could impact the precipitation cycle.
Later studies in the US and Europe would show that the highly acidic rainfall (i.e., “acid rain”) had a deleterious effect on lakes and various ecosystems (e.g., Beamish and Harvey 1972). Likens and Borrmann (1974) would show that acidic precipitation and particles were negatively impacting forests in the northeastern United States-specifically in the region in Fig. 9 that underlies the elongated “front” of high excess-\({\mathrm{SO}}_{4}^{2-}\) values that extend over the eastern US into New England.
Throughout his career, Junge continued to provide leadership to bring awareness of the importance of consistent cross-disciplinary research on the impact of anthropogenically-driven changes on atmospheric composition and climate (Junge 1975a, b, 1977; Junge et al. 1975).
3.9 The book Air Chemistry and Radioactivity (1963)
In 1963 Christian Junge published the book Air Chemistry and Radioactivity (Fig. 10). This book is a classic. It was the first to portray atmospheric chemistry as an integrated discipline with global-scale relevance, a concept that provided the comprehensive picture and vision that would catalyze the development of the field in subsequent decades. Indeed, for some 15 years after its publication, Junge's book was generally regarded as the "bible" of atmospheric chemistry. The book was instrumental in bridging the gap between classical chemistry and meteorology, two disciplines that had previously barely acknowledged each other.

The cover of Junge (1963) (With permission from Elsevier)
Junge pointed out in his Preface that in summarizing the field of atmospheric chemistry, sub-divisions “can be made on the basis of chemical compounds or their phases, and overlapping of chapters and sections is inevitable in both cases.” We note that even today authors of atmospheric chemistry books face this dilemma. For example, does one look at the sulfur cycle in its entirety in one section, or does one cover sulfur in gas, aerosol, and precipitation in separate chapters? Junge decided to sub-divide his book by phases, with chapters on the chemistry of gases, aerosols, and precipitation. He included a fourth chapter on radioactivity, which cut across all three phases. Finally, he included a very brief chapter on air pollution to the extent that the urban areas that generate air pollution are important sources of many of the chemicals that can also impact the non-urban atmosphere on larger scales.
In the chapter on gases, Junge addressed three research areas for each gas – its observed distribution in time and space, its sources and sinks, and its budget and cycle. In effect, this was a unique early attempt to develop a biogeochemical cycle for each gas. He pointed out that for most gases, information to address the aerosol and precipitation phases cited above was largely lacking. On the very first page of the book Junge points out the relationship between the variability of the concentration of a gas to its atmospheric residence time – a concept that he developed much more quantitatively in Junge (1974) and that is the topic of Sect. 3.3 above.
The chapter on aerosols contains sections on both the physical properties of aerosols and their chemical composition, as well as their distribution in the troposphere and stratosphere. The physical properties section has sub-headings on size distributions, sedimentation, and coagulation, as well as optical and electrical properties and the role of aerosols in water vapor condensation. The log radius-number distribution that he developed (see Sect. 3.2 above) is covered in this chapter. The section on the chemical composition of aerosols includes discussions on the nature and sources of Aitken particles as well as sea salt aerosols and continental aerosols. Finally, there is a section on the distribution of tropospheric and stratospheric aerosols.
Radioactivity in the atmosphere was particularly important at that time because of the extensive nuclear testing programs of several nations, resulting in the release of a variety of radionuclides into the atmosphere. Junge used the bomb nuclide data along with emissions from nuclear reactors, radioactive isotopes generated by cosmic rays, and natural emanations from the soils (e.g., radon-222 and its decay products) to improve his understanding of atmospheric circulation and removal processes for aerosols and gases. In the extensive chapter on atmospheric radioactivity, he has sections on the use of radioactivity from all those sources to elucidate environmental processes. These topics are important to understanding how radioactive debris might impact human health although this issue is not addressed in his book.
The chapter on the chemistry of precipitation addresses issues of wet and dry removal of aerosols and gases from the atmosphere as well as the chemical composition of precipitation. In the latter section, there is an extensive discussion of the composition of several chemical constituents of precipitation in various geographical regions, especially in the United States.
While this book was not written specifically as a textbook, it became mandatory reading for students of atmospheric chemistry in the 1960s and 1970s, and even later. Junge (1963) covers the literature through 1960 and roughly the first half of 1961, with over 500 references. Figure 11 shows the number of references used in the book for each year of publication from 1930 through mid-1961. This figure clearly shows the rapid increase in atmospheric chemistry publications (at least those referenced) after about 1950. It is interesting to compare that number with the more than one thousand journal publications on atmospheric chemistry each year today.

Reading again today his comprehensive and ground-breaking book of almost 400 pages, one can appreciate the insights Junge had over 60 years ago and his remarkable ability to proceed from small-scale studies to global views and visions. Junge states in the Preface to his book “… if this book helps to close the gap between meteorology, chemistry, and nuclear chemistry and helps to stimulate research in this field, the effort will have been worthwhile.” It certainly did that!
3.10 Exploring the budget of atmospheric mercury
One of the projects pursued within the “Collaborative Research Center” of the German Research Foundation (DFG) was focused on the cycle of atmospheric mercury. The project was initiated by Junge at the beginning of 1970s, and the first atmospheric measurements were made by C. Eberling onboard a ship in the northern Atlantic Ocean and from an aircraft primarily above the Pacific Ocean. These data were published as part of his Ph.D. dissertation (Eberling 1975) and in Seiler et al. (1980). To continue this work, Franz Slemr took a post-doctoral position at the Max Planck Institute for Chemistry in 1975. As an introduction he received from Junge a series of hand-written notes with Junge’s thoughts on the atmospheric mercury cycle. Some 66 pages of these notes still survive (see Online Resource). One part is dated December 5, 1976, so the notes are based on data from about a dozen publications before 1977. These are listed in the reference lists of Slemr et al. (1981, 1985).
In the mid-1970s there were only a few measurements of mercury concentrations in the atmosphere, precipitation, ocean, fossil fuels and wood. The analytical methods of those times allowed a reliable determination of total gaseous mercury and of particulate mercury in the atmosphere. Measurements of mercury, both elemental and compounds (Hg°, HgCl2, CH3HgCl2, (CH3)2Hg and related compounds) using specific absorption tubes were also reported (Braman and Johnson 1974; Johnson and Braman 1974), but the method proved unreliable for low concentrations in ambient air. Based on this extremely limited and partly erroneous data, including studies from his own group, Junge used his unique capability of combining sparse observations with the known physicochemical properties of mercury and its compounds and with the dispersion properties of the atmosphere to roughly estimate various Hg fluxes and to create an initial estimate of the global sources, sinks and the lifetime of atmospheric mercury. Although later measurements with improved analytical techniques did not confirm his estimates, the notes illustrate Junge’s way of thinking: always viewing a topic from several different angles. Based on the solubility of elemental mercury and organic mercury compounds in water he deduced that mercury emitted from the ocean surface must exist predominantly as elemental mercury. Using the same argument, he concluded that mercury found in precipitation consist of mercury compounds formed from elemental mercury by some atmospheric oxidation processes. He had already speculated about an oxidation by halogens in the stratosphere and by ozone and OH radicals in the troposphere. It is worth mentioning that because of uncertain measurements of mercury species in air (Jaffe et al. 2014) the question of mercury oxidants and their oxidation products is still waiting for its definitive answer.
The notes include one of the first tentative budgets for atmospheric mercury presented in Fig. 12. The translation of that page and associated explanatory material are presented in Table 2.

A tentative budget for atmospheric mercury prepared by Christian Junge in about 1976–1977, page 8 of Junge’s notes (From and with permission of Franz Slemr)
Notes concerning page 8 and material from relevant later papers
Page 8 of the notes provides the tentative estimate of mercury emissions prepared by Christian Junge in about 1976–1977 based on data that were available at that time. The current estimate of total emissions of mercury to the atmosphere is less than 1 × 1010 g yr−1 (Holmes et al. 2010; Horowitz et al. 2017). The tentative budget of Junge overestimated the emission primarily because the early mercury concentration measurements in water, soil, coal, etc. were significantly overestimated due to contamination during sampling and analysis. For example, Junge refers on page 5 of his notes to dissolved elemental mercury concentrations in seawater of 200 pg/l, while the currently reported concentrations are around 8 pg/l (Mason et al. 2017). The gradients of Hg in the atmosphere used by Junge were also not confirmed by later measurements (e.g., Weigelt et al. 2016). An enormous quantity of data on mercury concentrations and its behavior in the environment has been accumulated since Junge´s time (AMAP/UNEP 2019). Nonetheless, Junge’s budget provided a framework for subsequent critical experiments, the results of which led to an improved budget and a better understanding of atmospheric mercury (Slemr et al. 1981, 1985).
4 International leadership and honors
Because of Junge’s many abilities and accomplishments, he was often asked to accept important leadership positions in the international atmospheric chemistry and atmospheric sciences communities. From 1963 to 1975, he served as Secretary and then President of the international Commission on Atmospheric Chemistry and Radioactivity (currently the international Commission on Atmospheric Chemistry and Global Pollution) of the International Association of Meteorology and Atmospheric Physics (IAMAP). From 1975 to 1979 he served as President of IAMAP. Junge was the first atmospheric chemist to become President of IAMAP, now the International Association of Meteorology and Atmospheric Sciences (IAMAS). His election was a clear indication that atmospheric chemistry had become recognized as an integral and important part of the atmospheric sciences – a recognition that largely resulted from Junge’s own research and leadership. He also served as President of the Gesellschaft für Aerosolforschung (Association for Aerosol Research) from 1977 to 1978.
Major honors were bestowed on Junge because of his pioneering research and leadership. These include the following: Member of the Leopoldina Academy of Science in 1964; Fellow of the American Meteorological Society (AMS) in 1967; the Alfred-Wegener-Medal of the German Meteorological Association in 1968; the Carl-Gustav-Rossby Medal of the AMS in 1973; and the Korrespondierendes Mitglied der Mainzer Akademie der Wissenschaften und Literatur (Corresponding Member of the Mainz Academy of Science and Literature) in 1976. The University of Frankfurt honored him as Honorary Doctor of Natural Philosophy in 1978, and he became a Foreign Member of the American Academy of Arts and Sciences in the same year. In 1981 the German government honored him with “Das große Verdienstkreuz des Verdienstordens der Bundesrepublik Deutschland” (Grand Cross of Merit of the Federal Republic of Germany). The European Aerosol Association established the Junge Memorial Award in honor of Christian Junge in 1999. The Junge Award recognizes the outstanding research contributions of an individual who has developed a completely new area in aerosol science and/or technology, as Junge had done. The wording associated with the Alfred-Wegener-Medal is a particularly appropriate statement of Junge’s leadership in the international development of global-scale atmospheric chemistry: “with special appreciation of his unending endeavors to view his research results in the light of the entire meteorological phenomena and the general circulation.”
5 Memories and reflections
As mentioned earlier, Christian Junge is generally recognized as the founder of the modern discipline of atmospheric chemistry (sometimes described by him as “Air Chemistry”). He achieved this by taking disparate facts previously known about atmospheric composition (e.g., see Cauer 1951) and combining them with meteorology to develop conceptual and mathematical models. These would serve as strong foundations for research on global atmospheric chemistry. His scientific accomplishments have been described in previous sections. However, his success depended on more than intellectual insights. Two previous papers have discussed aspects of Junge’s life and science (Georgii 1978; Duce 1997), and the reader is referred particularly to the personal account of Georgii (1978), one of Junge’s first students.
Junge would often say that “successful science is a mixture of luck, competence, and hard work.” However, he brought more to science than these three attributes alone. Junge had universal interests; from understanding the tiniest particle, to the atmospheric chemistry of previous eons, and the chemistry of intergalactic space. He was happy to work across disciplinary boundaries while maintaining a strong respect for the scientific knowledge of his collaborators. Junge was generous with his knowledge. Contrary to the image of the remote grand-professor, he spent much time in mutual and nurturing discussion with young scientists from other institutions and he encouraged scientists in his own institute to develop their own scientific interests. Here we bring together some recollections of scientists who worked with Junge that shed light on his scientific life and his personal history.
Perhaps a sidelight, but in Fig. 13 is a photograph of a watercolor painting by Junge in 1946 which shows his love of the outdoors and nature. It is perhaps significant that in representing landscapes in paintings he emphasizes the broad features of the scene rather than the details, as was his approach to atmospheric chemistry. This painting hung in the office of one of Junge’s technicians at the German Weather Service long after his departure, until 1977 when Peter Winkler was given it rather than have it thrown out.

A watercolor painting by Christian Junge in 1946 (From and with permission of Peter Winkler
Ruprecht Jaenicke was initially a Ph.D. student of Junge and later rose to become a group leader under Junge. He recalls conversations with Junge in which Junge provided details of his early years in science.
“An early interest in flying and air traffic inspired Junge to study meteorology. He selected meteorology as a career rather than chemistry, which he liked, partly because he had seen unemployed chemists looking for work during the Great Depression of the late 1920s and 1930s. Seeking the best education, he enrolled in the University of Graz, Austria, because Alfred Wegener was a professor there, but after Wegener died in Greenland in 1930, he changed to universities in Hamburg and Frankfurt.”
Given his association with Wegener, it must have been a particular pleasure for Junge to receive the Alfred-Wegener-Medal of the German Meteorological Association in 1968 (see Sect. 4).
Ruprecht Jaenicke continues:
“The struggle of making a career was for Junge, like many of his generation, a difficult task. Seeing little chance for a successful career in Germany in the early 1950s, he accepted an invitation from Dr. Helmut Landsberg, Director of the Geophysics Directorate at the US Air Force Cambridge Research Laboratories in Bedford, Massachusetts, to join that research center. There he made multiple substantial contributions including one of his great discoveries, that of the stratospheric sulfate aerosol layer, now known as the “Junge layer” (see Sect. 3.1). Unfortunately, as the layer was not meteoric material that would be hazardous to satellites but relatively harmless sulfate particles that originated from emissions at the earth’s surface and not space, his research support was ended, and his position was terminated. He was given a year’s funding while he looked for a new position.”
This was a painful loss to Junge, but he went on to ultimately much better employment in Germany. This loss of employment at AFCRL occurred during a major reorganization and sharpened focus on military research (AFCRL 1962).
Bob Duce, while a graduate student at MIT in the early 1960s, was privileged to meet Christian Junge several times while Junge was at the Air Force Cambridge Research Laboratories. Bob writes:
“Junge’s clear insights, his patience, his sincere interest, and his desire to help a graduate student made a lasting impression. Junge also provided me with a set of page proofs for his 1963 book before it was published so I could take it with me to Hawaii, where I was undertaking fieldwork for my Ph.D. dissertation. Needless to say, those page proofs were devoured in Hawaii!”
Returning to Ruprecht Jaenicke’s account:
“When Junge returned to Germany in 1962, he became Professor of Meteorology at the University of Mainz. However, he found he was limited in attracting personnel because of the difficult staffing structure of German universities, and he had to overcome numerous difficulties. For example, colleagues from the chemistry faculty were not supportive and were reluctant to share space or recommend students. The only building initially available for his group was a renovated World War Two army barracks on an abandoned military airfield. In this building Junge’s own office was only 9 m2, and there was no conference room or pantry. Junge was also able to get funds from the Volkswagenstiftung to construct a temporary office building for 6 scientists. However, that building had serious problems. Each night the air in the building became saturated with formaldehyde from the plywood construction materials. Besides its negative health effects, it irritated the eyes. Every morning, the residents of this office space gathered at the building entrance like penguins to select who should go in first and open the windows! Another example was that, in an attempt to make the most of limited funding, Junge organized a field campaign on Tenerife Island with a holiday airline. They provided transport and housing by the sea, but later the university accountants did not want to reimburse him because they thought the travel was a holiday. Nonetheless, Junge overcame these difficulties, and he soon began to develop his broadscale research studies of atmospheric chemistry.”
“In 1968 Junge was appointed director at the Max Planck Institute (MPI) for Chemistry and formed the Department of Air Chemistry. He was initially reluctant to accept the appointment because he did not have a formal education as a chemist. However, when he became convinced that he could hire chemists instead, and he realized the huge opportunities he would have in such a position, he accepted. After all the earlier obstructions and distractions, Junge subsequently developed at the MPI for Chemistry the leading international research program in atmospheric chemistry.”
In 1972/1973 Ian Galbally made his first trip around the world to many institutes to learn about atmospheric chemistry and visited Mainz. He writes:
“Junge, the Director, started my day’s visit with what from memory was an hour-long conversation about atmospheric chemistry, including a break for mineral water and coffee. The discussion ranged across everything in atmospheric chemistry and to the fringes, including astrochemistry, which I knew nothing about but was keen to learn. The discussion also covered local history and the wooden pilings of a Roman bridge in the Rhine at Mainz. I then met the various groups in the Institute and learnt of their individual work.”
Junge could have a lighthearted approach to describing his science at times. He was initially convinced that the upper size of atmospheric aerosol particles should be around 20 µm and those particles should sediment out of the troposphere in about a day. He and Ruprecht Jaenicke undertook many studies around Germany to confirm such an upper limit, but they failed. Then Junge decided on the probability approach in a lighthearted way. Sean Twomey told Ian Galbally that when Junge was asked at a conference what the upper limit to the aerosol size distribution is, he replied that if you put an impactor on a mountainside, eventually an aircraft will fly into it.
As described in Sect. 3.10, in 1975 Franz Slemr took a post-doctoral position at the Max Planck Institute for Chemistry, having completed his Ph.D. studies there under Peter Warneck. His allocated post-doctoral project was to investigate the atmospheric mercury cycle. To introduce Slemr into a completely new subject, Junge gave him 66 pages of hand-written notes with his thoughts on the mercury cycle as noted briefly above (see Online Resource). Based on a few available measurements of mercury, Junge roughly estimated the global sources, sinks and lifetime of atmospheric mercury, providing a notion of what the atmospheric mercury cycle could look like. The notes illustrate Junge´s multifaceted thinking, which was characterized by combining meteorology with physical chemistry and sparse available measurements. These notes also provided the framework for Franz´s subsequent work. Franz says:
“The notes and Junge´s unassuming manner left their mark on me through my whole professional career”.
Peter Winkler worked with Christian Junge in the early days at the Max Planck Institute for Chemistry and writes:
“Junge approached new scientific questions as a generalist, and it was enough for him when he could identify new atmospheric chemical processes. Studying the details, he left to others; it was important to him if a process could be recognized as significant and could be described by a simple formula. He also sought to strengthen cooperation: international coordination was important for implementing new global atmospheric chemistry research programs. Junge’s position was that one did not need to be an expert to do good research, but that a promising approach and suitable method were sufficient to tackle open scientific questions. He therefore employed the geologist Manfred Schidlowski, with whom he subsequently worked on the paleoatmosphere to find out more about the conditions under which life could step over from water to land.
During the IAMAP meeting in Seattle in 1977, scientists became aware of the poorly understood middle atmosphere. Some groups demanded Junge, as the IAMAP president, define new research fields that should be reserved exclusively for special research groups. As a young participant, I was struck by Junge’s refusal. Junge argued that the quality of research cannot be guaranteed by ownership of a research area. Later, at that meeting, Junge gave a lecture on planetary atmospheres and on the insight studying them provided about the development of the earth's atmosphere. Freedom of research, he claimed, was the best tool for recognizing important processes in terms of their magnitude and the relevant parameters and progress in science.”
Peter Winkler also writes that Junge was not infallible in atmospheric chemistry:
“When speculations first came up about a possible decline of the ozone layer and the necessity to substantiate this assumption, he wouldn't support a respective research grant with the comment that the main reactions determining the ozone formation and destruction were already known and new research wouldn't be required.”
However, in spite of this event, Junge’s contribution to atmospheric chemistry was outstanding.
Cecilia Payne-Gaposchkin (1977), has written:
“The reward of the young scientist is the emotional thrill of being the first person in the history of the world to see something or to understand something. Nothing can compare with that experience. The reward of the old scientist is the sense of having seen a vague sketch grow into a masterly landscape.”
No doubt Junge enjoyed both pleasures as he laid the foundation for a quantitative understanding of atmospheric composition and chemistry, and lived to see a new generation of scientists, including his successors at the Max Planck Institute for Air Chemistry, Paul Crutzen, Director 1980–2000, Nobel Laurate 1995, and Jos Lelieveld, Director 2000-present, leading research that adds colorful details to the picture.
In retirement, Junge continued his quest for knowledge in new and interesting areas beyond those in science, including art, anthropology, and English history. Nonetheless, his interest in atmospheric chemistry remained strong. Junge continued as an Honorary member (and a past President) of the international Commission on Atmospheric Chemistry and Global Pollution; each year he was sent the meeting minutes to which he would reply with a card and short note of encouragement.
In a note to Joe Prospero about 5 years before Junge passed away, he expressed his concerns about aging, saying that as one grows older, one fears that one will become forgetful, but worse, that one will be forgotten. In conclusion, we hope that atmospheric scientists in general and atmospheric chemists in particular will never forget Christian Junge and the many fundamental and enduring contributions he made to our discipline as well as his mentoring, guidance and friendship that he provided to so many people engaged in it.
6 Postscript
This paper is a contribution by colleagues, most of whom knew Christian Junge. In the early years of atmospheric chemistry, the participation of female and minority scientists was scant, as attested by Fig. 2. We acknowledge that this is also reflected in the current author list. We are pleased and supportive that gender balance and minority involvement are progressing in our field, although there remains room for improvement at all levels especially among the senior ranks.
Data availability statement
Not applicable for this review paper.
References
AFCRL.: History and progress of AFCRL, Jan. 1961-June 1962: A detailed survey of research at the Air Force Cambridge Research Laboratories, Office of Aerospace Research, Bedford, Mass. 4(3), 212 (1962)
AMAP/UNEP.: Technical background report for the Global Mercury Assessment 2018 arctic monitoring and assessment programme Norway/UNEP chemicals and health branch, Geneva, Switzerland Oslo 978-82-7971-108-7, (2019)
Archibald, A.T., NeuJ, L., Elshorbany, Y.F., et al.: Tropospheric ozone assessment report: A critical review of changes in the tropospheric ozone burden and budget from 1850 to 2100. Elementa-Sci. Anthropocene. 8(1), 034 (2020). https://doi.org/10.1525/elementa.2020.034
Auer, R.: Über den täglichen Gang des Ozongehalts der bodennahen Luft. Beit. Zur Geophysik 55, 137–145 (1939)
Beamish, R.J., Harvey, H.H.: Acidification of the La Cloche Mountain Lakes, Ontario, and resulting fish mortalities. J. Fish Board of Canada 29, 1131–1143 (1972)
Bigg, E.K.: The detection of atmospheric dust and temperature inversions by twilight scattering. J. Meteorology 13, 262–268 (1956)
Braman, R.S., Johnson, D.L.: Selective adsorption tubes and emission technique for determination of ambient forms of mercury in air. Environ. Sci. Technol. 8, 996–1003 (1974)
Brühl, C., Lelieveld, J., Crutzen, P.J., Tost, H.: The role of carbonyl sulphide as a source of stratospheric sulphate aerosol and its impact on climate. Atmos. Chem. Phys. 12, 1239–1253 (2012)
Castleman, A.W., Jr., Munkelwitz, H.R., Manowitz, B.: Isotopic studies of the sulfur component of the stratospheric aerosol layer. Tellus 26(1–2), 222–234 (1974)
Cauer, H.: Some problems of atmospheric chemistry. Compendium of Meteorology, Amer. Met. Soc., Boston, 1126–1136 (1951)
Chagnon, C.W., Junge, C.E.: The vertical distribution of sub-micron particles in the stratosphere. J. Atmos. Sci. 18, 746–752 (1961)
Clark, W.E., Whitby, K.T.: Concentration and size distribution measurements of atmospheric aerosols and a test of the theory of self-preserving size distributions. J. Atmos. Sci. 24, 677–687 (1967)
Clifford, H.M., Spaulding, N.E., Kurbatov, A.V., et al.: A 2000 year Saharan dust event proxy record from an ice core in the European Alps. J. Geophys. Res. Atmospheres. 124, 12,882–12,900 (2019). https://doi.org/10.1029/2019JD030725
Crutzen, P.J.: Gas-phase nitrogen and methane chemistry in the atmosphere. University of Stockholm, Sweden (1973)
Crutzen, P.J.: The possible importance of CSO for the sulfate layer of the stratosphere. Geophys. Res. Lett. 3, 73–76 (1976)
Czeplak, G., Junge, C.: Studies of interhemispheric exchange in the troposphere by a diffusion model. In: Frenkiel, F. N., Munn, R. E. (eds.) Turbulent diffusion in environmental pollution. Academic Press, New York. Adv. Geophys. 18B, 57–72 (1974)
Darwin, C.: An account of the fine dust which often falls on vessels in the Atlantic Ocean. Quart J. Geol. Soc. 2, 26–30 (1846)
Delany, A.C., Delany, A.C., Parkin, D.W., Griffin, J.J., Goldberg, E.D., Reimann, B.E.F.: Airborne dust collected at Barbados. Geochim. Cosmochim. Acta 31, 885–909 (1967)
Duce, R.A.: The impact of atmospheric nitrogen, phosphorus, and iron species on marine biological productivity. In: Buat-Menard, P., (ed.) The Role of Air-Sea Exchange in Geochemical Cycling, pp 497–529. Reidel, Dordrecht (1986)
Duce, R.A.: Obituary for Prof. Christian Junge, EOS, Bulletin of the American Geophysical Union 78, 39–40 (1997)
Eberling, C.: Ein Beitrag zur Bestimmung von Quecksilber in ng/m3-Bereich in atmosphärischer Luft. PhD Thesis, University of Mainz (1975)
Editor: Project Shower, an investigation on warm rainfall in Hawaii. Tellus 9, 471 (1957)
Ehhalt, D.H., Rohrer, F., Wahner, A., Prather, M.J., Blake, D.R.: On the use of hydrocarbons for the determination of tropospheric OH concentrations. J. Geophys. Res. 103, 18981–18997 (1998)
Ehmert, A.: Chemical Ozone Measurement. Advances in Chemistry, ACS Series. 21, 128–132 (1959). https://doi.org/10.1021/ba-1959-0021.ch018
Eichmann, R., Ketseridis, G., Schebeske, G., et al.: n-Alkane studies in the troposphere. 1. Gas and particulate concentrations in North Atlantic air. Atmos. Environ. 13, 587–599 (1979). https://doi.org/10.1016/0004-6981(79)90187-2
Eichmann, R., Ketseridis, G., Schebeske, G., et al.: n-Alkane studies in the troposphere. 2. Gas and particulate concentrations in Indian-Ocean air. Atmos. Environ. 14, 695–703 (1980). https://doi.org/10.1016/0004-6981(80)90053-0
Fishman, J., Crutzen, P.J.: Origin of ozone in the troposphere. Nature 274, 855–858 (1978)
Galloway, J. N., Likens, G. E., Hawley, M. E.: Acid precipitation: Natural versus anthropogenic components. Science 226, 829–831 (1984). https://doi.org/10.1126/science.226.4676.829
Georgii, H. W.: Highlights of the research in atmospheric chemistry during the last 25 Years. Pageoph. 116, 215–2f21 (1978)
Ghan, S. J., Liu, X., Easter, R. C., et al.: Toward a minimal representation of aerosols in climate models: Comparative decomposition of aerosol direct, semidirect, and indirect radiative forcing. J. Climate 25, 6461–6476 (2012). https://doi.org/10.1175/JCLI-D-11-00650.1
Gibbs, A.G., Slinn, W.G.N.: Fluctuations of trace gas concentrations in the troposphere. J. Geophys. Res. 78, 574–576 (1973)
Götz, F. W. P., Volz, F.: Aroser Messungen des Ozongehalts der unteren Troposphäre und sein Jahresgang. Z. Naturforschung. 6a, 634–639 (1951)
Hamrud, M.: Residence time and spatial variability for gases in the atmosphere. Tellus 35B, 295–303 (1983)
Hellmann, G., Meinardus, W.: Der grosse Staubfall vom 9. bis 12. März 1901 in Nordafrika, Süd- und Mitteleuropa, Abh. Kgl. Preuss Met. Inst. 2, No. 1, pp 93 (1901)
Hofmann, D.J., Rosen, J.M.: On the background stratospheric aerosol layer. J. Atmos. Sci 38, 168–181 (1981)
Holmes, C.D., Jacob, D.J., Corbitt, E.S., et al.: Global atmospheric model for mercury including oxidation by bromine atoms. Atmos. Chem. Phys. 10, 12037–12057 (2010)
Horowitz, H.M., Jacob, D.J., Zhang, Y., et al.: A new mechanism for atmospheric mercury redox chemistry: implications for the global mercury budget. Atmos. Chem. Phys. 17, 6353–6371 (2017)
Huneeus, N., Schulz, M., Balkanski, Y., et al.: Global dust model intercomparison in AeroCom phase 1. Atmos. Chem. Phys. 11, 7781–7816 (2011). https://doi.org/10.5194/acp-11-7781-2011
Israel, H., Schulz, L.: Über die Größenverteilung der atmosphärischen Ionen. Meteorol. Z. 49, 226–233 (1932)
Jaenicke, R.: Physical aspects of the atmospheric aerosol. In: Georgi, H.W., Jaeschke, W. (eds.) Chemistry of the polluted and unpolluted troposphere, 341–373. D. Reidel, Norwell, MA (1982)
Jaenicke, R.: Die Erfindung der Luftchemie – Christian Junge. In Kant, H., Reinhardt, C. (eds) 100 Jahre Kaiser-Wilhelm-/Max-Planck-Institut für Chemie (Otto-Hahn-Institut). Facetten seiner Geschichte, pp 187–202. Archiv der Max-Planck-Gesellschaft, Berlin (2012)
Jaenicke, R., Davies, C.N.: The mathematical expression of the size distribution of atmospheric aerosols. J. Aerosol Sci. 7, 255–259 (1976)
Jaffe, D.A., Lyman, S., Amos, H.M., et al.: Progress on understanding atmospheric mercury hampered by uncertain measurements. Environ. Sci. Technol. 48, 7204–7206 (2014). https://doi.org/10.1021/es5026432
Jickells, T., Moore, C. M.: The importance of atmospheric deposition for ocean productivity. Annu. Rev. Ecol. Evol. Syst. 46, 481–501 (2015)
Jobson, B.T., McKeen, S.A., Parrish, D.D., et al.: Trace gas mixing ratio variability versus lifetime in the troposphere and stratosphere: Observations. J. Geophys. Res. 104, 16090–16113 (1999)
Johnson, D.L., Braman, R.S.: Distribution of atmospheric mercury species near ground. Environ. Sci. Technol. 8, 1003–1009 (1974)
Jones, I.T.N., Wayne, R.P.: The photolysis of ozone by ultraviolet radiation. IV. Effect of photolysis wavelength on primary step. Proc. R. Soc. A: Math. Phys. Eng. Sci. 319, 273–287 (1970)
Jordan, C. E., Pszenny, A. A. P., Keene, W. C., et al.: Origins of aerosol chlorine during winter over north central Colorado, USA. J. Geophys. Res. Atmos. 120, 678–694 (2015). https://doi.org/10.1002/2014JD022294
Junge, C.E.: Übersättigungsmessungen an atmosphärischen Kondensationskernen. Gerlands Beiträge Zur Geophysik 46, 108–129 (1935)
Junge, C.E.: Nuclei of atmospheric condensation. Compendium of Meteorology, Amer. Met. Soc., Boston, MA. 182–191 (1951)
Junge, C.E.: Gesetzmäßigkeiten in der Größenverteilung atmosphärischer Aerosole über dem Kontinent.". Berichte Des Deutschen Wetterdienstes in Der US-Zone 35, 261–277 (1952)
Junge, C.E.: Die Rolle der Aerosole und der gasförmigen Beimengungen der Luft im Spurenstoffhaushalt der Troposphäre. Tellus 5, 1–26 (1953)
Junge, C.E.: The chemical composition of atmospheric aerosols 1. Measurements at Round Hill Field Station, J. Meteorol. 11, 323–333 (1954)
Junge, C.E.: The size distribution and aging of natural aerosols as determined from electrical and optical data in the atmosphere. J. Meteorol. 12, 13–25 (1955)
Junge, C.E.: Recent investigations in air chemistry. Tellus 8, 127–139 (1956)
Junge, C.E.: Chemical analysis of aerosol particles and of gas traces on the Island of Hawaii. Tellus 9, 528–537 (1957)
Junge, C.E.: Atmospheric chemistry. Adv. Geophys. 4, 1–108 (1958)
Junge, C.E.: Air Chemistry. Bull. Amer. Met. Soc. 40, 493–498 (1959)
Junge, C.E.: Global ozone budget and exchange between stratosphere and troposphere. Tellus 14, 363–377 (1962a)
Junge, C.E.: Note on the exchange rate between the northern and southern atmosphere. Tellus 14, 242–246 (1962b). https://doi.org/10.3402/tellusa.v14i2.9543
Junge, C.E.: Air Chemistry and Radioactivity. Academic Press, New York and London (1963)
Junge, C.E.: Our knowledge of physico-chemistry of aerosols in undisturbed marine environment. J. Geophys. Res. 77, 5183–5200 (1972). https://doi.org/10.1029/JC077i027p05183
Junge, C.E.: Residence time and variability of tropospheric trace gases. Tellus 26, 477–488 (1974)
Junge, C.E.: Die Entstehung der Erdatmosphäre und ihre Beeinflussung durch den Menschen. Max-Planck-Gesellschaft Jahrbuch, 309–317 (1975a)
Junge, C.E.: Transport mechanisms for pesticides in the atmosphere. Pure Appl. Chem. 42, 95–104 (1975b)
Junge, C.E.: Basic considerations about trace constituents in the atmosphere as related to the fate of global pollutants. In: Suffet, L.H. (ed.) Fate of Pollutants in the Air and Water Environments, Part 1, vol. 8, pp. 7–23. John Wiley, New York (1977)
Junge, C.E., Chagnon, C.W., Manson, J.E.: Stratospheric aerosols. J. Meteorol. 18, 81–108 (1961a)
Junge, C.E., Chagnon, C.W., Manson, J.E.: A world-wide stratospheric aerosol layer. Science 133, 1478–1479 (1961b)
Junge, C.E., Czeplak, G.: Some aspects of seasonal variation of carbon dioxide and ozone. Tellus 20, 422–434 (1968). https://doi.org/10.3402/tellusa.v20i3.10021
Junge, C.E., Gustafson, P.E.: On the distribution of sea salt over the United States and its removal by precipitation. Tellus 9, 164–173 (1957)
Junge, C.E., Manson, J.E.: Stratospheric aerosol studies. J. Geophys. Res. 66, 2163–2182 (1961)
Junge, C.E., Robinson, E., Ludwig. F.L.: A study of aerosols in Pacific air masses. J. Appl. Met. 8, 340–347 (1969). https://doi.org/10.1175/1520-0450(1969)008<0340:ASOAIP>2.0.CO;2
Junge, C.E., Schidlowski, M., Eichmann, R., Pietrek, H.: Model calculations for terrestrial carbon cycle – carbon isotope geochemistry and evolution of photosynthetic oxygen. J. Geophys. Res. Oceans Atmos. 80, 4542–4552 (1975)
Junge, C.E., Werby, R.T.: The concentration of chloride, sodium, potassium, calcium, and sulfate in rainwater over the United States. J. Meteorol. 15, 417–425 (1958)
Keene, W.C., Gallway, J.N., Likens, G.E., et al.: Atmospheric wet deposition in remote regions: Benchmarks for environmental change. J. Atmos. Sci. 72, 2947–2978 (2015)
Keene, W.C., Pszenny, A.A.P., Galloway, J.N., et al.: Sea-salt corrections and interpretation of constituent ratios in marine precipitation. J. Geophys. Res. 91, 6647–6658 (1986). https://doi.org/10.1029/JD091iD06p06647
Ketseridis, G., Hahn, J., Jaenicke, R., Junge, C.E.: The organic constituents of atmospheric particulate matter. Atmos. Environ. 10, 603–610 (1976). https://doi.org/10.1016/0004-6981(76)90045-7
Krotkov, N.A., McLinden, C.A., Li, C., et al.: Aura OMI observations of regional SO2 and NO2 pollution changes from 2005 to 2015. Atmos. Chem. Phys. 16, 4605–4629 (2016)
Lelieveld, J., Dentener, F.J.: What controls tropospheric ozone? J. Geophys. Res. 105, 3531–3551 (2000)
Levy, H.: Normal atmosphere – large radical and formaldehyde concentrations predicted. Science 173, 141–143 (1971)
Likens, G.E., Bormann, F.H.: Acid rain: A serious regional environmental problem. Science 184, 1176–1179 (1974)
Linke, F.: Die Sonnenstrahlung und ihre Schwächung in der Atmosphäre. Handbuch Geophysik. 8, 239–338 (1942)
Long, M.S., Keene, W.C., Easter, et al.: Sensitivity of tropospheric chemical composition to halogen-radical chemistry using a fully coupled size-resolved multiphase chemistry/global climate system: Halogen distributions, aerosol composition, and sensitivity of climate-relevant gases. Atmos. Chem. Phys. 14, 3397–3425, (2014). https://doi.org/10.5194/acp-14-3397-2014
Mason, R.P., Hammerschmidt, C.R., Lamborg, C.H., et al.: The air-sea exchange of mercury in the low latitude Pacific and Atlantic Oceans. Deep-Sea Res. I(122), 17–28 (2017)
Mihalopoulos, N., Putaud, J.P., Nguyen, P.C., Belviso, S.: Annual variation of atmospheric carbonyl sulfide in the marine atmosphere in the southern Indian Ocean. J. Atmos. Chem. 13, 73–82 (1991)
Odén, S.: The Acidification of Air Precipitation and its Consequences in the Natural Environment. Bulletin of the Ecological Research Communications NFR. Arlington, VA:Translation Consultants Ltd. (1968)
Opik, E.J.: Interplanetary dust and terrestrial accretion of meteoritic matter. Irish Astron. J. 4, 84–135 (1956)
Payne-Gaposchkin, C.H.: Russell Prize Lecture of the American Astronomical Society – 50 years of novae. Astron. J. 82, 665–673 (1977)
Penndorf, R.: The vertical distribution of Mie particles in the troposphere. J. Atmos. Sci. 11, 245–245 (1954)
Peterson, J.T., Junge, C.E.: Sources of particulate matter in the atmosphere. In: Matthews, W.H. (ed.) Man’s Impact on the Climate, pp. 310–320. MIT Press, Cambridge, MA. (1971)
Prospero, J.M., Carlson, T.N.: Vertical and areal distribution of Saharan dust over the western equatorial North Atlantic Ocean. J. Geophys. Res. 77, 5255–5265 (1972)
Prospero, J.M., Delany, A.C., Delany, A.C., Carlson, T.N.: The discovery of african dust transport to the western hemisphere and the Saharan air layer: A history. Bull. Amer. Meteorol. Soc. 102, 1–53 (2021)
Prospero, J.M., Lamb, P.J.: African droughts and dust transport to the Caribbean. Climate Change Implications. Science 302, 1024–1027 (2003)
Regener, V.H.: The vertical flux of atmospheric ozone. J. Geophys. Res. 62, 221–228 (1957)
Saltzman, E.S., Savoie, D.L., Prospero, J.M., Zika, R.G.: Atmospheric methanesulfonic acid and non-sea-salt sulfate at Fanning and American Samoa. Geophys. Res. Lett. 12, 437–440 (1985). https://doi.org/10.1029/GL012i007p00437
Sanchez, K.J., Chen, C.L., Russell, L.M., et al.: Substantial seasonal contribution of observed biogenic sulfate particles to cloud condensation nuclei. Sci. Rep. 8, 3235 (2018). https://doi.org/10.1038/s41598-018-21590-9
Schultz, M.G., Akimoto, H., Bottenheim, J.W., et al.: The Global Atmosphere Watch reactive gases New Hampshire measurement network. Elem. Sci. Anth. 3, 000067 (2015). https://doi.org/10.12952/journal.elementa.000067
Schultz, M.G., Schroder, S., Lyapina, O., et al.: Tropospheric ozone Assessment report: Database and metrics data of global surface ozone observations. Elem. Sci. Anth. 5, 58 (2017). https://doi.org/10.1525/elementa.244
Schütz, L., Jaenicke, R.: Particle number and mass distributions above 10–4 cm radius in sand and aerosol of the Sahara Desert. J. Appl. Meteorol. 13, 863–870 (1974)
Seiler, W., Eberling, C.: Slemr, F: Global distribution of gaseous mercury in the troposphere. PAGEOPH 118, 964–974 (1980)
Slemr, F., Seiler, W., Schuster, G.: Latitudinal distribution of mercury over Atlantic Ocean. J. Geophys. Res. 86, 1159–1166 (1981)
Slemr, F., Schuster, G., Seiler, W.: Distribution, speciation, and budget of atmospheric mercury. J. Atmos. Chem. 3, 407–434 (1985)
Slinn, W.G.N.: A simple model for Junge’s relationship between concentration fluctuations and residence times for tropospheric trace gases. Tellus 40B, 229–232 (1988)
SMIC (Study of Man´s Impact on Climate) Report: Inadvertent Climate Modification: MIT Press, ISBN 9780262191012, pp 334 (1971)
Sofen, E.D., Bowdalo, D., Evans, M.J., et al.: Gridded global surface ozone metrics for atmospheric chemistry model evaluation. Earth Syst. Sci. Data. 8, 41–59, (2016). https://doi.org/10.5194/essd-8-41-2016
Stein, A.F., Draxler, R.R., Rolph, G.D., et al.: NOAA’s HYSPLIT Atmospheric Transport and Dispersion Modeling System. Bull. Amer. Meteor. Soc. 96, 2059–2077 (2015). https://doi.org/10.1175/BAMS-D-14-00110.1
Stohl, A., Hittenberger, M., Wotawa, G.: Validation of the Lagrangian particle dispersion model FLEXPART against large scale tracer experiment data. Atmos. Environ. 32, 4245–4264 (1998). https://doi.org/10.1016/S1352-2310(98)00184-8
Teichert, F.: Vergleichende Messung des Ozongehaltes der Luft am Erdboden und in 80 m Höhe. Z. Meteorol 9, 21–27 (1955)
Warneck, P.: Chemistry of the natural atmosphere, 2nd edn. Academic Press, San Diego, CA (2000)
Weigelt, A., Ebinghaus, R., Pirrone, N., et al.: Tropospheric mercury vertical profiles between 500 and 10 000 m in Central Europe. Atmos. Chem. Phys. 16, 4135–4146 (2016)
Whitby, E.R., McMurry, P.H.: Modal aerosol dynamics modeling. Aerosol Sci. Technol 27, 673–688 (1997)
Wikipedia: Stratospheric sulfur aerosols. (2022). https://en.wikipedia.org/wiki/Stratospheric_sulfur_aerosols. Accessed Aug 2021
Willeke, K., Whitby, K.T.: Atmospheric aerosols: size distribution interpretation. J. Air Poll. Control Assoc. 25, 529–534 (1975)
Williams, J., de Reus, M., Krejci, R., et al.: Application of the variability-size relationship to atmospheric aerosol studies: estimating aerosol lifetimes and ages. Atmos. Chem. Phys. 2, 133–145 (2002)
Williams, J., Fischer, H., Harris, G.W., et al.: The variability-lifetime relationship for organic trace gases: a novel aid to compound identification and estimation of HO concentrations. J. Geophys. Res. 105, 20473–20486 (2000)
Zhao, A., Ryder, C.L., Wilcox, L.J.: How well do the CMIP6 models simulate dust aerosols? Atmos. Chem. Phys. 22, 2095–2119 (2022)
Acknowledgements
We are pleased to acknowledge information provided by Hans Volkert. and William Fitzgerald as well as excellent advice and suggestions provided by reviewers Michael Prather and Peter Liss
Funding
Open Access funding enabled and organized by Projekt DEAL. The co-authors did not receive support from any organization for the submitted work.
Author information
Authors and Affiliations
Contributions
The following table identifies the primary authors of the various sections of this review paper. All co-authors contributed to the study’s conception and design. All co-authors contributed either text or comments to almost every section. All co-authors commented on previous versions of the manuscript and all read and approved the final manuscript.
Section | RAD | RRD | IEG | JNG | RJ | WCK | JL | HLII | JMP | LS | FS | PW |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
1 | X | X | X | |||||||||
2 | X | X | X | X | X | X | ||||||
3.1 | X | X | ||||||||||
3.2 | X | X | ||||||||||
3.3 | X | X | ||||||||||
3.4 | X | X | ||||||||||
3.5 | X | X | X | X | X | |||||||
3.6 | X | X | X | X | X | |||||||
3.7 | X | X | X | |||||||||
3.8 | X | X | X | X | X | |||||||
3.9 | X | |||||||||||
3.10 | X | X | X | |||||||||
4 | X | X | ||||||||||
5 | X | X | X | X | X | X | X | X |
Corresponding authors
Ethics declarations
Competing interests
The co-authors have no relevant financial or non-financial interests to disclose, and they have no competing interests to declare that are relevant to the content of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Duce, R.A., Dickerson, R.R., Galbally, I.E. et al. Christian Junge – a pioneer in global atmospheric chemistry. J Atmos Chem 79, 219–256 (2022). https://doi.org/10.1007/s10874-022-09437-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10874-022-09437-0