
Abstract
C-terminal binding proteins (CtBP1/2) are transcriptional coregulators that play a significant role during vertebrate neurodevelopment. This systematic review aims to identify case reports with genetic variants in CTBP1 and CTBP2 associated with brain development syndromes.
We screened different databases (PubMed, Scopus, Google Scholar, LILACS) by systematically searching journals and checking reference lists and citations of background papers. We found fourteen cases (10 males) from five papers carrying two pathogenic, heterozygous variants in the CTBP1 gene (13 individuals carried the missense mutation c.991C T, p.Arg342Trp, and one subject carrying the 2-base pair deletion c.1315_1316delCA, p.Gln439ValfsTer84). These mutations were de novo in 13 cases and one case of maternal germinal mosaicism. Two variants are in the same domain of the protein: Pro-Leu-Asp-Leu-Ser (PLDLS) C terminal. Patients with these mutations exhibit a phenotype with intellectual disability, HADDTS syndrome (hypotonia, ataxia, developmental delay, and tooth enamel defects), and cerebellar volume loss. We did not identify reported cases associated with homozygous mutations harbored in CTBP1. We did not identify any report of neurodevelopment phenotypes associated with heterozygous or homozygous CTBP2 mutations. Due to CTBP2/RIBEYE being a gene with dual function, identifying and interpreting the potential pathogenic variants is challenging.
Further, homozygous mutations in the CTBP2 gene may be lethal. The mechanisms involved in the pathogenesis of neurodevelopment due to variants of these proteins have not yet been elucidated, despite some functional evidence. Further studies should be conducted to understand these transcription factors and their interaction with each other and their partners.
Similar content being viewed by others
Introduction
C-terminal binding proteins (CTBP1 and CTBP2) are two highly conserved proteins expressed in different tissues of vertebrate species [3] and share 76% homology [4]. The primary function of the CTBP family members is to be a transcriptional corepressors. Since these proteins do not bind directly to DNA, they form a corepressor complex to perform their function by developing dimers with chromatin-modifying enzymes (histone deacetylases and methyltransferases), DNA-binding proteins, chromodomain-containing proteins, and CoREST proteins [5]. Other functions are controlling the equilibrium between tubular and stacked structures in the Golgi complex and brown adipose tissue differentiation.
CTBPs have three main domains: The substrate-binding domains, which contain the Pro-X-Asp-Leu-Ser (PXDLS) binding sequence, the central domain Arg-Arg-Thr (RRT), responsible for NAD(H) binding and dimerization, and a C-terminal domain. The partners of CTBPs are sequence-specific that bind to the PXDLS domain [2]. Though CTBP 1/2 share similar functions, they have some differences. The CTBP1 gene is on chromosome 4p, and CTBP2 is on chromosome 10q. Both proteins are ubiquitously expressed in all human tissues. However, CTBP2 appears to be expressed earlier in development. Only CTBP2 has a nuclear localization signal at its N-terminus. Conversely, CTBP1 has a PDZ-binding domain at its C-terminus for cytoplasmic functions with particular proteins such as neuronal nitric oxide synthase [6]. CTBPs have alternative splicing. CTBP2 has two dual functions with each type of isoform. The CTBP2 isoform has the function of a transcription factor. The isoform called RIBEYE is the main component of synaptic ribbons or specialized synapses. CTBPs can form homodimers or heterodimers necessary to carry out their functions [2], but this relevance is not fully known.
CTBP family members have been implicated in critical functions for neural development in various species, including drosophila, xenopus [4], mice [7], and avians [6]. CTBPs have been implicated in developing neural tube closure, forebrain, and hindbrain in murine [6, 7]. In humans, although CTBPs have precise functions in brain development, few studies have focused on the exact role. Most studies are focused on cancer due to the participation of these transcription factors in various functions associated with cell proliferation and apoptosis. With this review, we want to identify possible polymorphisms in CTBP 1/2 that have been associated with or suggested as gene candidates for phenotypes in the human nervous system.
Methods
Key question
Have cases been reported with genetic variants in CTBP1 or CTBP2 genes associated with neurological, neurodegenerative, or neurodevelopmental diseases?
Eligibility criteria
-
Types of studies: case reports and case series were included. No language, publication date, or publication status restrictions were imposed.
-
Types of participants: humans. No restriction by mode of inheritance or transmission, nor by the type of variant or classification.
-
Types of comparison/intervention: genetic variants (exon or intron) in C-terminal binding proteins (CTBP1/2), without sequencing or genetic analysis restriction.
-
Types of outcome measures: all reports of clinical cases diagnosed with neurological, neurodegenerative, or neurodevelopmental diseases, including neural tube defects.
Information sources and selection
Studies were identified by searching electronic databases: PubMed, Scopus, Google Scholar, and LILACS. Other sources were hand searching of genetic journals, preprint server Health Science Case Reports Research Network (https://www.ssrn.com/index.cfm/en/hscasereprn/); DECIPHER database (https://www.deciphergenomics.org/), checking reference lists and citations of background papers. The search end date was 09 Jun 2022.
Search methods for the identification of studies
The following search strategies were used:
-
1.
MEDLINE—PubMed
The PubMed search strategy used is available in Table 1. We used the following search terms: “nervous system development,” nervous system embryology,” “neurodevelopmental disorders,” “intellectual disability,” “neural tube defect,” “CTBP2,” “CTBP1,” “humans,” “RIBEYE,” “BARS protein,” “C-terminal Binding Protein,” “Brefeldin A-Ribosylated Substrate,” “case series study,” “genetic association studies,” and “case report.”
Table 1 PubMed search strategy The final searches were ((humans) AND (((((((neurodevelopmental disorders) OR (intellectual disability)) OR (central nervous system embryology)) OR (nervous system development)) OR (nervous system embryology)) OR (neural tube defect)) AND ((((((CTBP1) OR (CTBP2)) OR (RIBEYE)) OR (BARS protein)) OR (C-Terminal Binding Protein)) OR (Brefeldin A-Ribosylated Substrate)))) AND ((((case series study)) OR (genetic association studies)) OR (case report)).
-
2.
SCOPUS
The search was carried out in documents by keyword/title or abstract without any restriction or filter (Table 2). We used the following search terms: “nervous system development,” “nervous system embryology,” “neurodevelopmental disorders,” “intellectual disability,” “neural tube defect,” “CTBP,” and “C-Terminal Binding Protein.”
Table 2 Scopus search strategy The final searches were ((TITLE-ABS-KEY (CTBP)) OR (TITLE-ABS-KEY (“C-Terminal Binding Protein”))) AND ((TITLE-ABS-KEY (neurodevelopmental AND disorders) OR TITLE-ABS-KEY (nervous AND system AND embryology) OR TITLE-ABS-KEY (nervous AND system AND development) OR TITLE-ABS-KEY (intellectual AND disability) OR TITLE-ABS-KEY (neural AND tube AND defect))).
-
3.
LILACS (Latin American and Caribbean Health Sciences database)
The search was carried out in subject/title/abstract. The term used was “CTPB.”
-
4.
Google Scholar
We used the same search terms used in PubMed combined with Boolean connectors.
Data extraction and analysis
The title and the abstract initially selected the articles returned by the searches. We read the full text of pre-selected studies and included papers that met the above criteria. Finally, the articles selected for the review were checked to avoid duplicate published data.
Results
Selection of studies
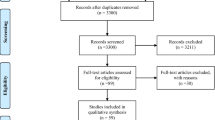
The search carried out to select the studies included in this review is detailed in Fig. 1. A total of 78 references were identified, with potentially valuable articles in PubMed = 7, Scopus = 21, and LILACS = 1. Google Scholar and hand searching were found an additional 49 studies. After adjusting for duplicate studies, 74 studies remained, which were screened by title and abstract. Of these, reports that did not meet the inclusion criteria were excluded, leaving us to review nine articles in full. In the analysis, five studies that met the inclusion criteria were included.

Flow diagram of study selection, following the PRISMA guidelines [8]
In DECIPHER database, a missense variant in CTBP2 is reported (c.979G > C, p.Glu327Gln), associated with an autism spectrum phenotype, cleft palate, diffuse white matter abnormalities, and severe intellectual disability. The variant was de novo and heterozygous. There is no published paper confirming the variant. In addition, the genotype of the reported individual appears associated with other additional variants in AUTS2 (c.3566 T > C, p.Leu1189Pro) and ITGB3 (c.985A > G, p.Asn329Asp) [9].
Characteristics of included studies
In our review, a total of 9 studies were identified in which a member of the CTBP family was involved. Within this search, there were studies reporting cases with distal chromosomal deletions on chromosomes 4p and 10q, where the syndrome was not specific for CTBP1 and CTBP2, respectively. Therefore, they were excluded from the phenotype analyses. Five included studies were summarized as shown in Table 3. Four excluded studies were summarized in Table 4.
Study design
We identify three case reports [11, 13, 14] and two case series reports [10, 12]. All five studies identified variants by whole-exome sequencing (WES). Sanger confirmed four reports and two studies with additional functional studies [11, 12].
Identified variants
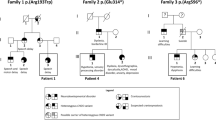
Two variants have only been reported in the CTBP1 gene. A variant (c.1315_ 1316delCA, p.Gln439ValfsTer84) has been reported in a single case, confirmed by Sanger but without functional studies [14]. The other 13 cases present the same recurrent heterozygous mutation (c.991CT, p.Arg342Trp). Beck et al. [10] report that case 1 presents another addition variant (in COL6A3) to CTBP1 with maternal somatic mosaicism. This study also reports that the mother of this same individual is healthy despite having this mosaicism.
Description of the cases
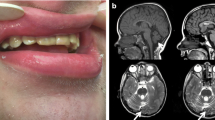
Fourteen individual cases and clinical characteristics were summarized in Tables 5 and 6. The nationality of the cases is not recorded in the publications. Eleven cases were described in the USA (cases 1–11, Table 4). One case was reported in the UK (case 12, Table 4), another in Iran (case 13, Table 4), and the last in India (case 14, Table 4). Severe intellectual disability (ID) or global development delay was present in twelve cases—eleven cases with significant gait disturbance, including 3 cases without gait. Cerebellar atrophy was identified in nine subjects. None of the cases reported seizures, except case 14, with a history of a single episode of myoclonus at 5 years of age. Three cases did not report defects in dental enamel.
Discussion
With this systematic review, we present evidence of five reports with 14 relatively homogeneous cases with a mutation in the CTBP1 gene. An additional study (the study by Bathia et al. [13]) was identified in this review, with a case not included in the clinical description by Khamirani et al. [14].
The phenotype of most cases includes developmental and language delay, intellectual disability, motor disturbance, muscle weakness, hypotonia, and cerebellar signs such as ataxia and dysarthria mainly, in addition to dental abnormalities and evidence of cerebellar and vermix atrophy. In some cases, cognitive, motor, and language regression were reported. A case of neurodegeneration and death at 16 years old.
The most-reported mutation (p.Arg342Trp) has been considered a recurrent mutation. Moreover, according to Kaplanis et al. [19], factors associated with recurrent mutations may be attributable to a verifiable phenotype in disease-causing mutations, which makes it easy to identify and report them. Another cause may also be increased mutability at the specific sites, and, finally, positive selection of mutations by “paternal age effect” and clonally expand over time [20]. Determining which factor influences more should be important for future studies. No reports mentioned the age of the parents; for example, developmental disorders caused by de novo mutations have been estimated to have an average prevalence at birth between 1 in 213 and 1 in 448, depending on the parents’ age [21].
Most of the individuals presented de novo mutation. This is quite common, mainly in rare diseases associated with neurological and psychiatric disorders such as intellectual disability, autism, and schizophrenia [22]. In case #14, the authors report the case as de novo mutation [14]. However, the parents are consanguineous, and a brother of the proband affected with a similar condition but not included in this analysis. Although parents were negative for the variant, this would indicate that it could be another mutation in another additional gene causing the disease. It was estimated that people with other affected family members were less likely to have de novo pathogenic mutations [21]. However, in the same study, it has been estimated that approximately 6% of individuals from consanguineous families have a probably pathogenic de novo mutation, which highlights the relevance of considering de novo causality in all families [21].
Of the cases reviewed here, 71% were male. A higher prevalence of autism spectrum disorder, DI, and attention deficit hyperactivity disorder have been observed in males [23, 24]. However, it has been found that women carry more pathogenic variants for brain development than men [25], and it has been observed that males have a 25% lower probability of being carriers of a probably pathogenic de novo mutation compared to females (OR = 0.75, 0.65–0.87 CI 95%) [21]. Thus, it has been considered a gender bias underlying phenotype or social bias [25].
Although reported cases represent highly penetrant alleles associated with single-gene disorders, mutations affecting domains important for protein interactions may also have subtle effects. Only heterozygous variants are found in this review. An autosomal dominant inheritance pattern would be present in family cases, with variable penetrance. CtBPs are coactivators or corepressors of transcription through interaction with other transcription factors and chromatin-modifying enzymes. Therefore, they are unable to bind to DNA independently. A proposed mechanism to explain Mendelian dominance in transcription factors is through a competitive binding [26]. There is competition between the transcription factor allelic variants for binding to the promoter sites they regulate. Nevertheless, this mechanism does not seem to apply to coregulators as CTBP family members.
Oligomerization is a critical factor for transcriptional activity in CTBPs, forming structures in dimers or tetramers by binding to the NAD(H) domain. These molecular complexes promote stability and interactions with DNA-binding factors [27]. Regarding the mechanism of CTBP1 mutation p.R342W to produce disease, a dominant negative effect has been proposed [12]. The complexes formed would be a mixture of mutated and wild-type subunits. The dominant negative effect would be more significant when more repeating subunits are included because the mutated subunits block the function of the wild-type molecules [28]. Other mechanisms could be additionally influencing. Mutations can perturb simply protein interactions, as shown by Beck et al. [12]. Another mechanism is stoichiometric imbalances when a certain amount of monomer increases in the complex [28].
We found no published papers with sequence variants in the CTBP2 gene. The cause of the absence of publications may be due to reduced penetrance and lethality of the mutation with increased prenatal or perinatal death (due to spontaneous abortion, termination of pregnancy due to a fetal anomaly, fetal death, or early neonatal death) [20]. CTBP2 homozygous null mice die early with brain malformations and axial truncations. This protein is necessary very early in development for exit from pluripotency and the formation of the three germinal layers of the embryonic stem cell [29]. CTBP2 has unique functions, but many other functions are shared with CTBP1 [7]. In addition, the CTBP2 isoform called RIBEYE has different functions in specialized neurons [30]. Variants in exons shared by both isoforms CTBP2/RIBEYE could be phenotypically masked and undetected [1].
The possible disease mechanism for CTBP1 mutation p.R342W seems still unclear despite functional evidence of the unstable association of several transcriptional regulatory proteins with the PXDLS-binding cleft, differences in the expression patterns of other genes involved in cellular pathways, and increased pro-apoptotic protein in fibroblasts from patients. The authors have hypothesized an alteration in neurodevelopment due to the absence of apoptotic regulation at the cerebellum level. Animal models with the variant could perhaps give new information. Furthermore, family genetic studies of inherited mutations could help to understand better these two fascinating transcription factors, the relationship between them, and the clinical implications associated with the interaction with their multiple partners.
References
Piatigorsky J (2001) Dual use of the transcriptional repressor (CtBP2)/ribbon synapse (RIBEYE) gene: how prevalent are multifunctional genes? Trends Neurosci 24(10):555–557. https://doi.org/10.1016/S0166-2236(00)01894-4
Quinlan KGR et al (2006) Role of the C-terminal binding protein PXDLS motif binding cleft in protein interactions and transcriptional repression. Mol Cell Biol 26(21):8202–8213. https://doi.org/10.1128/MCB.00445-06
Chinnadurai G (2007) Transcriptional regulation by C-terminal binding proteins. Int J Biochem Cell Biol 39(9):1593–607. https://doi.org/10.1016/j.biocel.2007.01.025
Turner J, Crossley M (2001) The CtBP family: enigmatic and enzymatic transcriptional corepressors. BioEssays 23(8):683–690. https://doi.org/10.1002/bies.1097
Shi Y et al (2003) Coordinated histone modifications mediated by a CtBP corepressor complex. Nature 422(6933):735–738. https://doi.org/10.1038/nature01550
Stankiewicz TR, Gray JJ, Winter AN, Linseman DA (2014) C-terminal binding proteins: central players in development and disease. Biomol Concepts 5(6):489–511. https://doi.org/10.1515/bmc-2014-0027
Hildebrand JD, Soriano P (2002) Overlapping and unique roles for C-terminal binding protein 1 (CtBP1) and CtBP2 during mouse development. Mol Cell Biol 22(15):5296–5307. https://doi.org/10.1128/MCB.22.15.5296-5307.2002
Page MJ et al (2021) The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ 372:n71. https://doi.org/10.1136/bmj.n71
Firth HV et al (2009) DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am J Hum Genet 84(4):524–533. https://doi.org/10.1016/j.ajhg.2009.03.010
Beck DB et al (2016) A recurrent de novo CTBP1 mutation is associated with developmental delay, hypotonia, ataxia, and tooth enamel defects. Neurogenetics 17(3):173–178. https://doi.org/10.1007/s10048-016-0482-4
Sommerville EW et al (2017) De novo CTBP1 variant is associated with decreased mitochondrial respiratory chain activities. Neurol Genet 3(5). https://doi.org/10.1212/NXG.0000000000000187
Beck DB et al (2019) A pathogenic CtBP1 missense mutation causes altered cofactor binding and transcriptional activity. Neurogenetics 20(3):129–143. https://doi.org/10.1007/s10048-019-00578-1
Bhatia SK, Arora V, Verma IC (2020) A further case of hypotonia, ataxia, developmental delay and tooth enamel defect syndrome due to a recurrent C-terminal binding protein 1 mutation. Clin Dysmorphol :148–151. https://doi.org/10.1097/MCD.0000000000000321
Khamirani HJ et al (2021) Exome sequencing identified a de novo frameshift pathogenic variant of CTBP1 in an extremely rare case of HADDTS. J Genet 100(2):1–5. https://doi.org/10.1007/s12041-021-01315-0
Shimizu K et al (2014) Microarray and FISH-based genotype-phenotype analysis of 22 Japanese patients with Wolf-Hirschhorn syndrome. Am J Med Genet Part A 164(3):597–609. https://doi.org/10.1002/ajmg.a.36308
Callaway D et al (2018) Prioritization of candidate genes for congenital diaphragmatic hernia in a critical region on chromosome 4p16 using a machine-learning algorithm. J Pediatr Genet 07(04):164–173. https://doi.org/10.1055/s-0038-1655755
Irving M et al (2003) Deletion of the distal long arm of chromosome 10: is there a characteristic phenotype? A report of 15 de novo and familial cases. Am J Med Genet 123A(2):153–163. https://doi.org/10.1002/ajmg.a.20220
Vera-Carbonell A et al (2015) Clinical comparison of 10q26 overlapping deletions: delineating the critical region for urogenital anomalies. Am J Med Genet Part A 167(4):786–790. https://doi.org/10.1002/ajmg.a.36949
Kaplanis J et al (2020) Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature 586(7831):757–762. https://doi.org/10.1038/s41586-020-2832-5
Goriely A, Wilkie AOM (2012) Paternal age effect mutations and selfish spermatogonial selection: causes and consequences for human disease. Am J Hum Genet 90(2):175–200. https://doi.org/10.1016/j.ajhg.2011.12.017
(2017) Prevalence and architecture of de novo mutations in developmental disorders. Nature 542(7642):433–438. https://doi.org/10.1038/nature21062
Gauthier J, Rouleau GA (2012) De novo mutations in neurological and psychiatric disorders: effects, diagnosis and prevention. Genome Med 4(9):71. https://doi.org/10.1186/gm372
Fombonne E (2009) Epidemiology of pervasive developmental disorders. Pediatr Res 65(6):591–598. https://doi.org/10.1203/PDR.0b013e31819e7203
Abikoff HB et al (2002) Observed classroom behavior of children with ADHD: relationship to gender and comorbidity. J Abnorm Child Psychol 30(4):349–359. https://doi.org/10.1023/a:1015713807297
Jacquemont S et al (2014) A higher mutational burden in females supports a “female protective model” in neurodevelopmental disorders. Am J Hum Genet 94(3):415–425. https://doi.org/10.1016/j.ajhg.2014.02.001
Porter AH, Johnson NA, Tulchinsky AY (2017) A new mechanism for Mendelian dominance in regulatory genetic pathways: competitive binding by transcription factors. Genetics 205(1):101–112. https://doi.org/10.1534/genetics.116.195255
Jecrois AM et al (2021) Cryo-EM structure of CtBP2 confirms tetrameric architecture. Structure 29(4):310-319.e5. https://doi.org/10.1016/j.str.2020.11.008
Bergendahl LT et al (2019) The role of protein complexes in human genetic disease. https://doi.org/10.1002/pro.3667
Kim TW, Kwak S, Shin J, Kang BH, Lee SE, Suh MY, Kim JH, Hwang IY, Lee JH, Choi J, Cho EJ, Youn HD (2017) Ctbp2-mediated β-catenin regulation is required for exit from pluripotency. Exp Mol Med 49(10):e385. https://doi.org/10.1038/emm.2017.147
Schmitz F, Königstorfer A, Südhof TC (2000) RIBEYE, a component of synaptic ribbons: a protein’s journey through evolution provides insight into synaptic ribbon function. Neuron 28(3):857–872. https://doi.org/10.1016/s0896-6273(00)00159-8
Acknowledgements
Study funded by the Universidad de Antioquia and MINCIENCIAS 1115-807-63223.
Funding
Open Access funding provided by Colombia Consortium
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Acosta-Baena, N., Tejada-Moreno, J.A., Arcos-Burgos, M. et al. CTBP1 and CTBP2 mutations underpinning neurological disorders: a systematic review. Neurogenetics 23, 231–240 (2022). https://doi.org/10.1007/s10048-022-00700-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-022-00700-w