
Abstract
The bacteria Vibrio cholerae causes cholera, an acute diarrheal infection that can lead to dehydration and even death. Over 100,000 people die each year as a result of epidemic diseases; vaccination has emerged as a successful strategy for combating cholera. This study uses bioinformatics tools to create a multi-epitope vaccine against cholera infection using five structural polyproteins from the V. cholerae (CTB, TCPA, TCPF, OMPU, and OMPW). The antigenic retrieved protein sequence were analyzed using BCPred and IEDB bioinformatics tools to predict B cell and T cell epitopes, respectively, which were then linked with flexible linkers together with an adjuvant to boost it immunogenicity. The construct has a theoretical PI of 6.09, a molecular weight of 53.85 kDa, and an estimated half-life for mammalian reticulocytes in vitro of 4.4 h. These results demonstrate the construct’s longevity. The vaccine design was docked against the human toll-like receptor (TLR) to evaluate compatibility and effectiveness; also other additional post-vaccination assessments were carried out on the designed vaccine. Through in silico cloning, its expression was determined. The results show that it has a CAI value of 0.1 and GC contents of 58.97% which established the adequate expression and downstream processing of the vaccine construct, and our research demonstrated that the multi-epitope subunit vaccine exhibits antigenic characteristics. Additionally, we carried out an in silico immunological simulation to examine the immune reaction to an injection. Our results strongly suggest that the vaccine candidate on further validation would induce immune response against the V. cholerae infection.
Similar content being viewed by others

Introduction
Cholera, which can cause very serious acute watery diarrhea and is also a symptom of social inequality and a lack of development, continues to pose a threat to public health which has cause emergency and brought to notice of the scientists. Dunkin (2021) and Nezafat et al. (2016) assert that cholera is a highly contagious disease that is spread by ingesting water or food that has been contaminated with the Vibrio cholerae bacterium. Cholera outbreaks have been documented during the previous 60 years in a number of African and Asian countries in 2021 and 2022. According to reports, there are sizable outbreaks still going on in Afghanistan, Bangladesh, the Democratic Republic of the Congo, Ethiopia, and Nigeria. Since the latest update on February 16, 2022, about 30,629 suspected cholera cases, including 39 fatalities, have been recorded globally. According to Ali et al. (2015), cholera cases range from 1.3 to 4.0 million each year, and the disease kills 21,000 to 143,000 people worldwide. However, 5–10% of people who have severe cholera cases—which are 50% deadly if untreated—are affected.
There have been over 287 suspected cholera cases reported in Nigeria, with 12 deaths. As of February 27, 2022, a total of 701 probable cases have been recorded from 12 states and Federal Capital Territory (FCTs), with 19 fatalities (CFR 2.7%). Five states, Taraba (242 cases), Cross River (111 cases), Borno (91 cases), Bayelsa (76 cases), and Adamawa, account for 82% of all cases. The age group most likely to be affected by the suspected cases are children under the age of 5 (WHO 2022). Even if the prevalence of cholera is not well known, it is nevertheless a serious tropical illness that is worth studying (CDC 2020; NORD 2021).
According to Somboonwit et al. (2017), not all strains of V. cholerae are considered to be the cause of cholera. Out of the more than 200 serology groups of V. cholerae, only the O1 and O139 groups have resulted in cholera epidemics (Waldor and Mekalanos 1994). Vaccination is a way to control and prevent cholera epidemics in the underdeveloped countries (Helen and Shoubai 2020) . Some of the known virulence factors of Vibrio cholerae include V. cholera toxin (CTX), toxin-coregulated pili (TCP), lipopolysaccharide (LPS), and outer membrane proteins (Omps), which are important candidates for the cholera vaccine (Nezafat et al. 2016).
Additionally, studies on their adverse consequences have been conducted (María et al. 2017). Also, additional research demonstrates that the multi-epitope vaccination has antigenic components that can elicit an immunological reaction (Validi et al. 2018; Khan et al. 2019; ul Qamar et al. 2021; Jyotisha and Qureshi 2022). This approach hereby eliminates some evaluations that have suggested that additional antigens may likely cause inflammation which are necessary for the immunogenicity of innovative vaccines in addition to identifying every antigen that may be investigated using conventional methods (Coscolla et al. 2015; Parvizpour et al. 2020).
In order to promote immunity and elicit both cellular and humoral immune responses to combat the cholera sickness, this study developed a novel multi-epitope peptide vaccine encompassing the cytotoxic T lymphocyte (CTL), helper T lymphocyte (HTL), and linear B lymphocyte (LBL) epitopes employing immune-informatics techniques in the development process. Using an in silico simulation, it was demonstrated that the constructed vaccine is capable of inducing immunological upon injection. The vaccine’s effectiveness and safety were assessed using physicochemical, molecular docking, and thermodynamic stability profiling approaches. Our study lays the groundwork for the experimental creation of a potent cholera vaccine.
Materials and methods
Retrieval of Vibrio cholerae polyprotein
In this research work, the best potential candidate protein sequences of Vibrio cholerae, O1 and O139 strain, were retrieved from NCBI Protein Database (https://www.ncbi.nlm.nih.gov/) in FASTA format for constructing multi-epitope vaccine by the use of immune-informatics approach (Dong et al. 2020). The protein sequences used were partial sequences (Shahab et al. 2022).
Antigenicity and allergenicity prediction
Antigenicity prediction was carried on the retrieved sequences using VaxiJen 2.0 webserver (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html (Abraham Peele et al. 2021). The server carries out the prediction of antigenic and non-antigenic amino acid sequences based on physicochemical properties only at a default threshold of 0.4 (Doytchinova and Flower 2007; Zaharieva et al. 2017; Habib 2020). All the sequences that passed the antigenicity test were further subjected to AlgPred webserver (https://webs.iiitd.edu.in/raghava/algpred/submission.html), an online tool to determine the allergenicity. The server uses six different approaches for the allergenicity prediction with 85% accuracy at 0.4 thresholds (Saha and Raghava 2006).
Cytotoxic T lymphocyte (CTL) epitope prediction
Cytotoxic T lymphocyte’s prediction of V. cholerae antigenic proteins were carried out using an online webserver- NetCTL (http://www.cbs.dtu.dk/services/NetCTL/) (Banerjee et al. 2020). The threshold was set to 0.75 to carry out the CTL epitopes prediction (Stranzl et al. 2010; Larsen et al. 2007).
Helper T lymphocyte (HTL) epitope prediction
Helper T lymphocyte’s prediction was carried out on the antigenic protein sequences using IEDB tool (http://www.iebd.org/immunogenicity/) according to the method of Vita et al. (2019). The human leukocyte antigen class II (HLA-DR) supertype alleles (DRB1*03:01, DRB3*01:01, DRB3*02:01, DRB3*02:02, DRB5*01:01) were selected, and three different methods were used in the prediction of the HLA-II epitopes; they are Consensus (smm/nn/sturniolo), Consensus (comb.lib./smm/nn), and NetMHCIIpan. The peptide affinity for each of the receptors is based on the IC50 score given to each and every predicted epitope. The same server was used for immunogenicity prediction as well, using major histocompatibility complex (MHC) class I immunogenicity (Vita et al. 2015).
Prediction of B cell epitopes
The B cell epitopes were predicted with an online tool server, BCpred (http://ailab-projects1.ist.psu.edu:8080/bcpred/SimpleServlet), and all parameters were left at default. The peptides, scores, and start position were shown by the predicted B cells (Chen et al. 2007).
Toxicity prediction
The predicted CTL, HTL, and B cell epitopes were submitted to ToxinPred, an online web server (http://crdd.osdd.net/raghava/toxinpred/) for toxicity prediction. The server classifies the epitopes as toxic and non-toxic based on their physicochemical properties (Gupta et al. 2013).
Construction of multi-epitope vaccine sequence
The predicted CTL, HTL, and B cell epitopes, sorted out through the discussed immune-informatics approach were used to design the vaccine construct. The adjuvant used was APPHALS, then EAAK linkers were used to link the adjuvant with the CTL epitope. The CTL epitope was linked with the 8 HTL epitopes using GPGPG linkers, while the 14 B cell epitopes were linked to the HTL epitopes with GPGPG linkers (Nagamune 2001).
Population coverage prediction
The epitopes selected for the Vibrio cholerae vaccine design must be predicted for their ability to binds to HLA molecules across different human population and the likelihood of it inducing long-lasting immune response in this population. IEDB population coverage tool was used in the prediction of the epitopes with high percentage of been presented in the context of HLA molecules to induce immune response in population (Bui et al. 2006). The endemic regions are selected for the population coverage.
Allergenicity prediction of the vaccine
Allergenicity prediction of the constructed vaccine was done using AlgPred (http://www.imtech.res.in/raghava/algpred/). The server used six different approaches for the prediction with 85% accuracy at 0.4 thresholds (Saha and Raghava 2006).
Antigenicity prediction of the vaccine
VaxiJen (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) was used for antigenicity prediction of the vaccine construct with bacteria selected as a model organism at a threshold of 0.4. The prediction of antigenic and non-antigenic amino acid sequences was carried out based on physicochemical properties only (Doytchinova and Flower 2007).
Physicochemical property prediction
ProtParam web tool (http://web.expasy.org/protparam/) was used for the examination of the physicochemical properties of the constructed sequence. Evaluation of the constructed amino acid sequence, the vaccine’s amino acid composition, in vivo/in vitro half life, theoretical pI, GRAVY (grand average of hydropathy) instability index, and molecular weight were all predicted (Gasteiger et al. 2005).
Secondary structure prediction for the vaccines
Secondary structure prediction of the constructed vaccine sequence was done using PSIPRED tool (http://bioinf.cs.ucl.ac.uk/psipred/), an online web-based server that performs secondary structure prediction given accurate result (Jones 1999).
Tertiary structure prediction
Tertiary structure prediction of the constructed sequence for the vaccine was carried out using I-TASSER (https://zhanggroup.org/I-TASSER/). The server generated 5 models, and the model with the highest C-score was selected (Yang and Zhang 2015) as predicted and validated by Zhou et al. (2022) to also model small protein structure.
Tertiary structure refinement
The final vaccine 3D structure was refined using Galaxy Refine tools (http://galaxy.seoklab.org/refine) (Adero et al. 2021). For the repacking of the amino acid sequence and side chain reconstruction, the server uses CASP10 refinement method. Also, the same method is employed for molecular dynamic simulation to relax the 3D structure of the query protein.
Molecular docking of vaccine
ClusPro 2.0 (https://cluspro.org), an online bioinformatics sever, was utilized for the docking of the vaccine with human TLR-4 accessed through PD ID of 4G8A from Pubchem (Ezediuno et al. 2021; Sanami et al. 2021). Twenty-six models were generated in respect to low energy, and large size was selected as it indicated a good interaction between the receptor and ligand.
In silico cloning and codon optimization of vaccine protein
The codon optimization was significant for the expression of vaccine structure in a host E. coli (strain K12) as the usage of a codon is comparatively different in E. Coli than the native host. Rho-independent transcription, restriction cleavage sites, and prokaryotic ribosome-binding sites were eluded by considering three extra options. Java codon adaptation tools provide the output in terms of CAI (codon adaptation index) and GC content in order to confirm the high level of protein expression. For cloning, the final vaccine construct in E. coli pET-28a ( +) vector modification N- and C-terminal with Xhol and Notl restriction sites were performed, respectively. Finally, for expression, the prepared optimized sequence, along with the restriction sites were incorporated into the pET-28a ( +) vector utilizing the SnapGene tool (https://www.snapgene.com) (Grote et al. 2005).
Immune simulation
To understand the molecular dynamics of the human immune system in response to designed vaccine, the relationship between the human immune system and vaccine construct was predicted using C-ImmSim 10.1 (https://kraken.ian.rm.cnr.it/C-IMMSIM/index.php?page=1), a server that uses agent-based modeling (Rapin et al. 2010). PSSM method was used to assess the production of cytokines and other substances like interferon and antibodies. Also, the response for T helper cell 1 and T helper cell 2 (Th1 and Th2) was predicted with the parameters left at default to measure the diversity or Simps Index by the server.
Results
Retrieval of antigenic proteins, antigenicity, and allergenicity prediction
A hundred (100) protein sequences of Vibrio cholerae were retrieved from the NCBI database (https://www.ncbi.nlm.nih.gov/) out of which only 46 passed the antigenicity and allergenicity analysis. Five structural polyproteins toxin-coregulated pili (TCPA and TCPF), outer membrane protein (OMPW and OMPU), and cholera toxin B subunit (CTB) of Vibrio cholerae with an antigenic probability score of greater than 0.8 were considered further for vaccine construction.
Cytotoxic T lymphocyte epitopes prediction
From all the 46 protein sequences that passed antigenicity and allergenicity analysis, a total of 290 epitopes were generated, out of which 195 were toxin-coregulated pili and others were cholera toxin subunit. However, based on MHC class 1 binding affinity, toxicity, allergenicity, and immunogenicity, only one CTL epitope as shown in Table 1 was selected; this is due to the fact that other epitopes did not pass the allergenicity test.
T lymphocyte epitopes prediction
Five reference sets of human alleles which includes HLA-DRB1*03:01, HLA-DRB3*01:01, HLA-DRB3*02:01, HLA-DRB3*02:02, and HLA-DRB5*01:01 were used for the prediction. A total of 230 HTL epitopes were subjected to the same predictions as CTL epitopes, in which only 8—208–222 (HLA-DRB5*01:01), 93–87 (HLA-DRB3*01:01), 248–262 (HLA-DRB3*02:01), 88–102 (HLA-DRB1*03:01), 84–98 (HLA-DRB1*03:01), 83–97 (HLA-DRB1*03:01), 130–144 (HLA-DRB1*03:01), and 85–99 (HLA-DRB1*03:01)—epitopes passed the predictions, and these were used for the designing of the vaccine (Table 2).
Prediction of B cell epitope
Out of the five cholera group of proteins, only 14 TcpA protein passed the allergenicity and toxicity test. These 14 linear B cell epitopes were used for the construction of multi-epitope vaccine using BCpred web server (http://ailab-projects1.ist.psu.edu:8080/bcpred/SimpleServlet) with the defaulted parameters. The predicted B cell epitopes shows (Table 3) the peptides, scores, and start position.
Construction of multi-epitope peptide vaccine
The final vaccine construct was designed by the combination of adjuvant and epitopes through the use of some linkers. 1 CTL, 8 HTL, and 14 B cells were made use in the designing of the subunit vaccine construct. With the use of EAAAK linker, the adjuvant was fused with the CTL epitope at the N-terminal; 8 HTL and 14 B cells were fused through the use of GPGPG linkers Figs. 1, 2 and 3.
Population coverage
The population coverage of the CTL epitopes across the endemic regions in Africa using the references HLA-A, HLA-B, and HLA-C alleles and that of HTL epitopes across the same regions using the references, HLA-DP, HLA-DQ, and HLA-DP selected during the MHC binding predictions are depicted in Table 4, while Fig. 4 is showing the percentage population coverage of the combined MHC molecules.

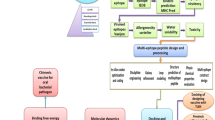
Systematic flow diagram for cholera vaccine construct. It is a flow chart of all the key steps involved in creating potential T cell, B cell, and multi-epitope vaccines against cholera. The details of the various steps are described in this section

Construction process of the vaccine

Epitope population coverage of the designed vaccine in endemic region of Africa

The peptide structure of the vaccine
Prediction, validation and refinement of 3D structure
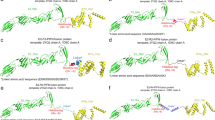
I-TASSER was used for the construction of the 3D structure. Five models were generated, out of which the best was selected based on the C-scores, the model with high C-score was selected because C-score of a higher value signifies a model with a higher confidence and vice versa. For refinement of the vaccine construct, Galaxy Refinement server was used in which five refined model were given but the best of them all was chosen based on RMSD, Mol Probity, High GDT-HA score, poor rotamers, and Rama favored. Model 5 3D structure is provided below. The model structure was chosen based on its structure information such as GDT-HA score of 0.9437, RMSD score of 0.450, Mol Probity calculated as 2.259, low clash score of 15.7, poor rotamers calculated as 0.5, and the Rama favored estimated to be 89.5 which is the highest and it is far better than other models of the vaccine, as shown in Table 5. Therefore, model 5 was selected as a refined vaccine structure out of the five refined models for further analysis. The 3D model of the vaccine selected was then subjected to structure assessment stage, and these includes ProCheck for protein model showing that 76.1% of amino acid are plotted in the most favorable region, 18.8% in allowed regions, and 2.7% in disallowed region. Also, ProSA-web and ERRANT estimated and verified the whole quality of the basic 3D model. The overall quality calculated by ERRANT was 64.02% and ProSA-web displayed a score of − 7.88 for the 3D model of the vaccine construct, which authenticate the precision of the vaccine. Result form ProSA-web and RAMPAGE are given in Fig. 5.

3D structure refinement, evaluation, and prediction. a The use of rampage Ramachandran plot for quality assessment of the vaccine model; the black square denotes torsion angle distribution relative to the core (red) and allowed (yellow) regions. Residues are plotted in generously allowed (pale yellow) and disallowed (white) regions. b In ProsaWeb, the prosa plot score of the vaccine can be identified as a black dot in the plot of all PDB structures’ quality scores of the same size. c ProsaWeb showing sequence position
Antigenicity and allergenicity assessment of the vaccine construct
Vaxijen and AlgPred servers were used for the prediction of antigenicity and allergenicity of the vaccine sequences, respectively, at a threshold of 0.4 for bacteria; the antigenicity server confirms that the candidate vaccine is antigenic with a score of 0.7844 and with the nearest protein in UniProtKB accession number Q86UU0, defined the vaccine as non-allergen on AlgPred.
Physiochemical properties of vaccines
The physiochemical properties of the designed vaccine sequences were predicted via ProtParam. Theoretical isoelectric point (theoretical PI) was predicted to be 6.09, molecular weight as 53.85 kDa, the estimated half-life for in vitro for mammalian reticulocytes is 4.4 h, while for in vivo in yeast and Escherichia coli is > 20 and > 10, respectively. The vaccine sequence aliphatic index was computed to be 60.32, which indicate the thermostability of the vaccine, and was classified as stable as the instability index (II) is computed to be 13.99 and the hydrophilic nature of the vaccine sequence was shown as the negative grand average of hydropathicity (GRAVY) was − 0.435.
Secondary structure prediction
In the secondary structure prediction of the vaccine construct which was carried out by PSIPRED, alpha-helix (Hh) was computed as 18.06%, extended strand (Ee) as 18.81%, and random coil (Cc) as 58.29%. Graphical representation of the secondary structure elements is shown in Fig. 6.

Graphical representation of the secondary structure of the vaccine construct
Molecular docking of constructed vaccine with human-toll like receptor-4
TLR-4 having PDB ID of 4G8A was selected as the receptor of choice for docking the vaccine which was carried out through a bioinformatics web server known as ClusPro 2.0 server. Twenty-six models were generated, but the best of it all in respect to low energy and large size was selected as it indicates a good interaction between the receptor and ligand. Graphically, docked complex intermolecular conformation and chemical interactions are presented below (Fig. 7).

Molecular docking of the vaccine with toll-like receptor-4. a Ribbon-like top-ranked model of docking assay. b Cartoon-like top-ranked model of docking assay
Immune simulation of the multi-epitope vaccine
Through in silico immune simulation, the injection of vaccine doses, the human immune system response was observed. The antibody titer was very high after the injection, and this triggered the immune response. IgM + IgG titer was observed at 700,000 antigens count per ml. IgM was around 400,000 xx/ml and IgG1 was reported as 250,000 xx/ml. Interleukin and cytokine responses were also observed,while IFN-gamma and IL-2 titer were increased significantly. All these indicate the dependability and high immune-triggering response upon injection. It also shows that there is high cellular immune response and memory cells’ development to pathogen recognition at re-occurrence. The population of the T cells, dendritic cells, phagocytic macrophages, and phagocytic natural killer cells were reported to be > 1150 cell/mm3, 225 cell/mm3, 225 cell/mm3, 374 cell/mm3, respectively. Therefore, all these results depicted in Fig. 8 denote that our vaccine candidate can trigger immune response effectively Fig. 9.

In silico immune simulation by the vaccine followed by injection. The immune response to the vaccine is demonstrated by antibodies titer. a Interferon and interleukins. b Natural killer cells. c Macrophages. d Cytotoxic T cell. e Dendritic cells. f Antigen and immunoglobulin

Codon adaptation using E. coli K12 strain
Codon adaptation and insilico cloning
Vaccine sequence design was modified using JCAT in reference to the E. coli K12 strain. The CAI value obtained was 0.1, and the GC content was 58.97%, indicating a greater level of vaccine design expression in the E. coli K12 strain. According to reports, GC levels between 35 and 70% have greater gene expression. The designed vaccine was cloned into the Pet-28a ( +) plasmid using the restriction cloning tool SnapGene. To do this, suitable restriction enzymes Xhol and Notl were selected, and their restriction sites were inserted at both ends of the modified nucleotide sequence (Habib 2020). The restriction sites for Xhol and Notl were added to the N- and C-termini of the optimized vaccine design and cloned in the pET-28a ( +) vector as shown in Fig. 10.

Cloned vaccine using Pet-28a( +) vector
Discussion
The use of vaccines has greatly improved the world’s health over a long period of time (Oli et al. 2020). In the past, developing vaccinations was thought to be the most practical method of preventing infectious diseases (Greenwood 2014). Herd immunity greatly reduces disease load, death, and disability (Helen and Shoubai 2020). Personalized vaccination is required for infectious organisms with complicated life cycles and antigenic diversity, and newly developing and re-emerging infectious diseases (ERID) bring additional challenges in vaccine development (Poland et al. 2016; Oli et al. 2020). With immunological findings and understanding of computational methods for epitope predictions, a new pattern of vaccine design has been revealed (Adhikari et al. 2018). Immune-informatics approaches are quick and efficient methods for discovering viable candidates for vaccine design using a pathogen’s proteome (Khan et al. 2019). According to Oladipo et al. (2022), epitope-based vaccine design is advantageous, profitable, extremely stable, non-toxic, and easy to engineer in comparison to conventional vaccines, which may raise several issues in patients who are compromised in immune system which also agrees with Wang et al. (2022) on the use of nucleic based vaccine. In silico immune-informatics methods can be used to accurately predict B and T cell epitopes using a plethora of bioinformatics tools, according to Farhadi et al. (2015).
This can have an impact on the formulation of epitope vaccines. Utilizing highly immunogenic and precise B cell and T cell epitopes that are extracted from the pathogen’s proteome, these multi-epitope-based vaccination peptides are made (Sajjad et al. 2020). Therefore, this method can not only identify all antigens that can be investigated using conventional approaches, but it can also identify additional antigens that are crucial to the immunogenicity of novel vaccines (Flower et al. 2010).
Consequently, cholera caused 2.8 million illnesses and 91,000 fatalities in more than 50 endemic nations. However, according to the most recent data, there were 69 different countries’ total of 2.9 million illnesses and 95,000 fatalities (Ali et al. 2015). It is highly desirable to develop an efficient and reliable cholera epitope vaccination. The current study’s objectives were to generate a cholera vaccine to lower worldwide mortality, and the findings will be validated by prior research in the field using the same approach with Banerjee et al. (2020). The majority of cholera vaccines now on the market or in development are inactivated (like Dukoral1, mORCAX1, and SancholTM) and live attenuated (like CVD103-HgR and Peru-15 or CholeraGarde1) (Holmgren 2021).
Nezafat et al. (2016) proposed that using immunostimulatory adjuvant (CTB) in epitope vaccine development, with the selection promiscuous epitopes from various V. cholerae protective antigens (OmpW, OmpU, TcpA, and TcpF) that bind to different HLA-II supertype alleles, and linking the epitopes with suitable linkers (EAAAK and GPGPG) to each other are important strategies to create CTL, HTL, and B cell epitopes were predicted using a variety of bioinformatics methods. The closest UniProt number, Q86UU0, which is designated as a non-allergen, antigenicity, and immunogenicity of synthetic peptides, was used to evaluate the allergenicity using the AlgPred service (Sharma et al. 2021; Adam 2021; Ghandadi 2022).
The immunization remained stable, nevertheless, as evidenced by the vaccine construct’s molecular weight of 53.85 Da and predicted PI of 6.09. The vaccine construct’s molecular weight of 53.85 Da and Pi of 6.09 show that it was stable. Cholera epitopes of various protective antigens (Ompu, Ompw, TcpA, and TcpF) that bind to different HLA-II supertype alleles with suitable linkers (EAAAK, GPGPG) are integrated into immune-stimulating extra vesicular CTB combined with CTB (Nezafat et al. 2016; Wieczorek et al. 2017).
To generate more precise epitopes that are limited to particular HLA-II supertype alleles, three servers (IEBD, SVMHC, and PSIpred) were employed, each with a different technique (Nielsen et al. 2010, 2020). B cell epitope is also necessary for the immunization to boost antibody response. The antigens ompU, ompw, TcpA, and tcpF were used to represent the frequently occurring area between mixed epitopes and B cell epitopes.
Because of its dual role in the formation of the epitope vaccination, the later region of the antigen was chosen. First, it limits the emergence of junctional epitopes (neoepitopes), and second, it improves immune processing and displays HLA-II binding epitopes (Livingstone et al. 2002). This area was joined using short amino acid linkers, particularly the GPGPG linker. To prevent contact with other vaccine segments, helical linkers were introduced to the N- and C-terminals, which improved the separation. CTB is a non-toxic subunit that acts as a mucosal immunostimulatory adjuvant, stimulating both mucosal and systemic immune responses (Stratmann 2015; Lavelle and Ward 2022). The population coverage of the designed cholera vaccine show that the designed cholera vaccine can induce immune response in an average of about over 60% of African population that cholera has been found to be endemic.
I-TASSER was used to simulate the initial 3D structure of the protein vaccine which has been reported to also modelled 3D of small protein sequences as validated by (Zhou et al. 2022). The model has to be modified, according to the results of the ProSA-web plot analysis of the preliminary version (Yang and Zhang 2015). After loop refinement and energy reduction of the core model, a high-quality 3-D standard necessary for the identification of conformational B cell epitopes and docking techniques was attained.
The CTB region of the vaccine and TLR2 were successfully docked by Cluspro (Kozakov et al. 2017). Due to CTB’s strong sequence and structural similarity to heat-labile enterotoxin B (LTB) and TLR2’s functional characteristics, an adequate docking model may be achieved even though there is no direct evidence of interaction between CTB and TLR2. The immunological and physicochemical characteristics of the protein vaccine were assessed (Dey et al. 2014). The vaccine is antigenic and also non-allergenic, according to our findings. Additionally, the vaccine demonstrated the appropriate level of solubility when it was overexpressed. The protein vaccines has high aliphatic index and high thermal stability. Finally, by modifying the DNA codon for production in the E. coli host and adding limit sites to gene flanks, the epitope vaccine was cloned in silico suggesting it a good candidate vaccine for Vibrio cholerae infection, and therefore, there is need for the use of wet laboratory techniques for further validation of the in silico prediction.
Conclusion
In our study, we used an immune-informatics approach to design a potential multi-epitope vaccine using six different groups of protein from vibrio cholerae. This cholera vaccine has the potential to provide prophylactic benefits. However, this in silico work requires experiment validation for confirmation, which will be quickly followed by in vitro and in vivo research to ensure the immunogenicity, wholesomeness, and safety of the potential vaccine.
Data availability statement
All data analyzed in this study are publicly available data at the National Centre for Biotechnology Information GenBank repositories (https://www.ncbi.nlm.nih.gov) and which could be made available upon request.
References
Abraham Peele K, Srihansa T, Krupanidhi S, Ayyagari VS, Venkateswarulu TC (2021) Design of multi-epitope vaccine candidate against SARS-CoV-2: a in-silico study. J Biomol Struct Dyn 39(10):3793–3801
Adam KM (2021) Immunoinformatics approach for multi-epitope vaccine design against structural proteins and ORF1a polyprotein of severe acute respiratory syndrome coronavirus-2 (SARS-CoV- 2). Tropical Diseases, Travel Medicine and Vaccines 7(1):1–13
Adero W, Nyanjom S, Mwirigi MK, Yattinder B (2021) In-silico epitope prediction of small ruminant morbillivirus proteins as potential vaccine candidates. East African Agricultural and Forestry Journal 85(1–4):14–14
Adhikari UK, Tayebi M, Rahman MM (2018) Immunoinformatics approach for epitope-based peptide vaccine design and active site prediction against polyprotein of emerging oropouche virus. J Immunol Res 2018
Ali M, Nelson AR, Lopez AL, Sack D (2015) PLoS Negl Trop Dis 9(6):e0003832. https://doi.org/10.1371/journal.pntd.0003832
Banerjee S, Majumder K, Gutierrez GJ, Gupta D, Mittal B (2020) Immuno-informatics approach for multi-epitope vaccine designing against SARS-CoV-2. BioRxiv
Bui HH, Sidney J, Dinh K, Southwood S, Newman MJ, Sette A (2006) Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinformatics 7:153. https://doi.org/10.1186/1471-2105-7-153
CDC (2020) Cholera: Vibrio cholerae infection. Centers for Disease Control and Prevention. Cole Supe´rieure de Biotechnologie de Strasbourg, Boulevard Se´bastien Brandt, 67400 Illkirch, France. Biologicals (2001) 29:209–213. https://doi.org/10.1006/biol.2001.0308, http://www.idealibrary.com
Chen J, Liu H, Yang J, Chou KC (2007) Prediction of linear B-cell epitopes using amino acid pair antigenicity scale. Amino Acids 33(3):423–428. https://doi.org/10.1007/s00726-006-0485-9
Coscolla M, Copin R, Sutherland J, Gehre F, de Jong B, Owolabi O, Gagneux SM (2015) Tuberculosis T cell epitope analysis reveals paucity of antigenic variation and identifies rare variable TB antigens. Cell Host Microbe 18(5):538–548
Dey AK, Malyala P, Singh M (2014) Physicochemical and functional characterization of vaccine antigens and adjuvants. Expert Rev Vaccines 13(5):671–685
Dong R, Chu Z, Yu F, Zha Y (2020) Contriving multi-epitope subunit of vaccine for COVID-19: immunoinformatics approaches. Front Immunol 11:1784
Doytchinova IA, Flower DR (2007) VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics 8(1):1–7
Dunkin MA (2021) Cholera. WebMD. https://www.webmd.com/a-to-z-guides/cholera-faq
Ezediuno LO, Onile OS, Oladipo EK, Majolagbe ON, Jimah EM, Senbadejo TY (2021) Designing multi-epitope subunit vaccine for ocular trachoma infection using Chlamydia trachomatis polymorphic membrane proteins G. Informatics in Medicine Unlocked 26:100764
Farhadi T, Nezafat N, Ghasemi Y, Karimi Z, Hemmati S, Erfani N (2015) Designing of complex multi-epitope peptide vaccine based on omps of Klebsiella pneumoniae: an in-silico approach. Int J Pept Res Ther 21(3):325–341
Flower DR, Macdonald IK, Ramakrishnan K, Davies MN, Doytchinova IA (2010) Computer aided selection of candidate vaccine antigens. Immunome Research 6(Suppl 2):S1. https://doi.org/10.1186/1745-7580-6-S2-S1
Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A (2005) In John M. Walker (ed): The Proteomics Protocols Handbook, Humana Press pp.571–607 Full text - Copyright Humana Press
Ghandadi M (2022) An Immunoinformatic strategy to develop new Mycobacterium tuberculosis multi-epitope vaccine. Int J Pept Res Ther 28(3):1–14
Greenwood B (2014) The contribution of vaccination to global health: past, present and future. Philosophical Transactions of the Royal Society B: Biological Sciences 369(1645):20130433
Grote A, Hiller K, Scheer M, Münch R, Nörtemann B, Hempel DC, Jahn D (2005) JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res 33(Web Server issue):W526–W531. https://doi.org/10.1093/nar/gki376
Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Open Source Drug Discovery Consortium and Raghava GP (2013) In silico approach for predicting toxicity of peptides and proteins. PLoS One 8(9):e73957. https://doi.org/10.1371/journal.pone.0073957
Habib PT (2020) Learning from COVID-19 Pandemic: In Silico Vaccine and Cloning Design Against Nipah Virus by Studying and Analyzing the Whole Nipah Virus Proteome
Helen HM, Shoubai C (2020) Advances in Vaccines. Current Application of Pharmaceutical Biotechnology 171:155–188. https://www.cdc.gov/cholera/infection-sources.html
Holmgren J (2021) An update on cholera immunity and current and future cholera vaccines. Tropical Medicine and Infectious Disease 6(2):64
Jones DT (1999) Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292(2):195–202. https://doi.org/10.1006/jmbi.1999.3091
Jyotisha SS, Qureshi IA (2022) Multi-epitope vaccine against SARS-CoV-2 applying immunoinformatics and molecular dynamics simulation approaches. J Biomol Struct Dyn 40(7):2917–2933
Khan M, Khan S, Ali A, Akbar H, Sayaf AM, Khan A, Wei DQ (2019) Immunoinformatics approaches to explore Helicobacter Pylori proteome (Virulence Factors) to design B and T cell multi-epitope subunit vaccine. Sci Rep 9(1):1–13
Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, Vajda S (2017) The ClusPro web server for protein–protein docking. Nat Protoc 12(2):255–278
Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M (2007) Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics 8(1):1–12
Lavelle EC, Ward RW (2022) Mucosal vaccines—fortifying the frontiers. Nat Rev Immunol 22(4):236–250
Livingston B, Crimi C, Newman M, Higashimoto Y, Appella E, Sidney J, Sette A (2002) A rational strategy to design multiepitope immunogens based on multiple Th lymphocyte epitopes. J Immunol 168(11):5499–5506
María RR, Arturo CJ, Alicia JA, Paulina MG, Gerardo AO (2017) The impact of bioinformatics on vaccine design and development. Vaccines 2:3–6
Nagamune T (2001) Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng Des Sel 14(8):529–532. https://doi.org/10.1093/protein/14.8.529
Nezafat N, Karimi Z, Eslami M, Mohkam M, Zandian S, Ghasemi Y (2016) Designing an efficient multi-epitope peptide vaccine against Vibrio cholerae via combined immunoinformatics and protein interaction-based approaches. Comput Biol Chem 62:82–95
Nielsen M, Lund O, Buus S, Lundegaard C (2010) MHC class II epitope predictive algorithms. Immunology 130(3):319–28. https://doi.org/10.1111/j.1365-2567.2010.03268.x. Epub 2010 Apr 12. PMID: 20408898; PMCID: PMC2913211
Nielsen M, Andreatta M, Peters B, Buus S (2020) Immunoinformatics: predicting peptide–MHC binding
NORD (2021) Rare disease https://rarediseases.org/rare-diseases/cholera/
Oladipo EK, Jimah EM, Irewolede BA, Folakanmi EO, Olubodun OA, Adediran DA, Akintibubo SA, Odunlami FD, Olufemi SE, Ojo TO, Akinro OP, Hezekiah OS, Olayinka AT, Abiala GA, Idowu AF, Ogunniran JA, Ikuomola MO, Adegoke HM, Idowu UA, Akindiya OE, Adelusi TI (2022) Immunoinformatics design of multi-epitope peptide for the diagnosis of Schistosoma haematobium infection. J Biomol Struct Dyn 1–8. Advance online publication. https://doi.org/10.1080/07391102.2022.2111358
Oli AN, Obialor WO, Ifeanyichukwu MO, Odimegwu DC, Okoyeh JN, Emechebe GO, Ibeanu GC (2020) Immunoinformatics and vaccine development: an overview. ImmunoTargets and Therapy 9:13
Parvizpour S, Pourseif MM, Razmara J, Rafi MA, Omidi Y (2020) Epitope-based vaccine design: a comprehensive overview of bioinformatics approaches. Drug Discov Today 25(6):1034–1042
Poland GA, Whitaker JA, Poland CM, Ovsyannikova IG, Kennedy RB (2016) Vaccinology in the third millennium: scientific and social challenges. Curr Opin Virol 17:116–125. https://doi.org/10.1016/j.coviro.2016.03.003
Rapin N, Lund O, Bernaschi M, Castiglione F (2010) Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 5(4):e9862
Saha S, Raghava GP (2006) Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 65(1):40–48. https://doi.org/10.1002/prot.21078
Sajjad R, Ahmad S, Azam SS (2020) In silico screening of antigenic B-cell derived T-cell epitopes and designing of a multi-epitope peptide vaccine for Acinetobacter nosocomialis. J Mol Graph Model 94:107477
Sanami S, Azadegan-Dehkordi F, Rafieian-Kopaei M, Salehi M, Ghasemi-Dehnoo M, Mahooti M, Bagheri N (2021) Design of a multi-epitope vaccine against cervical cancer using immunoinformatics approaches. Sci Rep 11(1):1–15
Shahab M, Hayat C, Sikandar R, Zheng G, Akter S (2022) In silico designing of a multi-epitope vaccine against Burkholderia pseudomallei: reverse vaccinology and immunoinformatics. J Genet Eng Biotechnol 20(1):1–12
Sharma N, Patiyal S, Dhall A, Pande A, Arora C, Raghava GPS (2021) AlgPred 2.0: an improved method for predicting allergenic proteins and mapping of IgE epitopes. Brief Bioinform 22(4):bbaa294. https://doi.org/10.1093/bib/bbaa294. PMID: 33201237
Somboonwit C, Menezes LJ, Holt DA, Sinnott JT, Shapshak P (2017) Current views and challenges on clinical cholera. Bioinformation 13(12):405
Stratmann T (2015) Cholera Toxin Subunit B as Adjuvant--An Accelerator in Protective Immunity and a Break in Autoimmunity. Vaccines (Basel) 3(3):579–96. https://doi.org/10.3390/vaccines3030579. PMID: 26350596; PMCID: PMC4586468
Stranzl T, Larsen MV, Lundegaard C, Nielsen M (2010) NetCTLpan: pan-specific MHC class I pathway epitope predictions. Immunogenetics 62(6):357–368. https://doi.org/10.1007/s00251-010-0441-4
ul Qamar MT, Ahmad S, Fatima I, Ahmad F, Shahid F, Naz A, Chen LL (2021) Designing multi-epitope vaccine against Staphylococcus aureus by employing subtractive proteomics, reverse vaccinology and immuno-informatics approaches. Comput Biol Med 132:104389
Validi M, Karkhah A, Prajapati VK, Nouri HR (2018) Immuno-informatics based approaches to design a novel multi epitope-based vaccine for immune response reinforcement against Leptospirosis. Mol Immunol 104:128–138
Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR, Wheeler DK, Sette A, Peters B (2019) The immune epitope database (IEDB): 2018 update. Nucleic Acids Res 47(D1):D339–D343. https://doi.org/10.1093/nar/gky1006
Vita R, Overton JA, Greenbaum JA et al (2015) The immune epitope database (IEDB) 3.0. Nucleic 449 acids research. 43:D405-D412
Waldor MK, Mekalanos JJ (1994) ToxR regulates virulence gene expression in non-O1 strains of Vibrio cholerae that cause epidemic cholera. Infect Immun 62(1):72–78
Wang L, Geng J, Chen L, Guo X, Wang T, Fang Y, Belingon B, Jiao Wu, Li M, Zhan Y, Shang W, Wan Y, Feng X, Xianghui Li Hu, Wang (2022) Improved transfer efficiency of supercharged 36 + GFP protein mediate nucleic acid delivery. Drug Deliv 29(1):386–398
Wieczorek M, Abualrous ET, Sticht J, Álvaro-Benito M, Stolzenberg S, Noé F, Freund C (2017) Major histocompatibility complex (MHC) class I and MHC class II proteins: conformational plasticity in antigen presentation. Front Immunol 8:292
World Health Organization (2022) Disease Outbreak News; Cholera – Malawi. Available at: https://www.who.int/emergencies/disease-outbreak-news/item/2022-DON419
Yang J, Zhang Y (2015) Protein structure and function prediction using I-TASSER. Curr Protoc Bioinformatics 52(1):5–8
Zaharieva N, Dimitrov I, Flower D, Doytchinova I (2017) Immunogenicity prediction by VaxiJen: a ten-year overview. J Proteomics Bioinformatics 10:298–310
Zhou X, Zheng W, Li Y, Pearce R, Zhang C, Bell, EW, Zhang G, Zhang Y (2022) I-TASSER-MTD: a deep-learning-based platform for multi-domain protein structure and function prediction. Nat Protoc 17(10):2326–2353. https://doi.org/10.1038/s41596-022-00728-0
Acknowledgements
The authors appreciate the staffs and management of Helix Biogen Institute for providing technical assistance during the cause of the work.
Author information
Authors and Affiliations
Contributions
EKO: Conceptualization, experimental design. OEA, GMA, FOB, ROA, HOA, KTK, JAO, GAA: Data retrieval, analysis, and wrote the first draft of the manuscript. SEO, TOO, OPA, OSH, ATO: Data analysis and result interpretation. EMJ, BAI, AFA, MOI, HMA, UAI: Result interpretation. EOF, OAO, DAA, SAA, FDO, OPO, OOB, SBA, MOB: Manuscript review and editing; all authors participated in the review of the final edition of the manuscript.
Corresponding author
Ethics declarations
Competing of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Oladipo, E.K., Akindiya, O.E., Oluwasanya, G.J. et al. Bioinformatics analysis of structural protein to approach a vaccine candidate against Vibrio cholerae infection. Immunogenetics 75, 99–114 (2023). https://doi.org/10.1007/s00251-022-01282-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-022-01282-5