
Abstract
The crystal structures of 8-R1-7-bromo-3-tert-butyl-1-R2-pyrazolo[5,1-c][1,2,4]triazin-4(1H)-ones 1a–c, 2a,c (R1 = CN, CO2Et, NO2, R2 = H, 1:1 and 3:1 solvates with DMSO; R1 = CN, CO2Et, R2 = CH2Boc), 8-R1-7-bromo-3-tert-butyl-1-R2-1,4-dihydropyrazolo[5,1-c][1,2,4]triazin-4-ols 3a,b (R1 = CN, R2 = n-Bu; R1 = Br, R2 = CH2Boc), 1,4-dihydro- and aromatic 7-R3-3-tert-butyl-4-R4-8-methylpyrazolo[5,1-c][1,2,4]triazines 5a,b, 6 (R3 = H, R4 = n-Pr; R3 = Br, R4 = n-Bu) were investigated by X-ray diffraction analysis. The structural preferences and different packing modes based on the intermolecular interactions were analyzed by the Hirshfeld surface and energy framework analysis.
Graphical Abstract
The crystal structures of ten 3-tert-butyl-4-oxo, 4-hydroxy- and 4-alkyl-7-bromopyrazolo[5,1-c][1,2,4]triazines including non-solvated, 1:1 and 3:1 solvates with DMSO were investigated by single crystal X-ray diffraction, Hirshfeld surface and energy framework analyses.

Similar content being viewed by others


Introduction
The chemistry of heterocyclic compounds is among the most extensively studied areas of the modern science [1, 2]. Nitrogen-containing fused systems are of particular research interest, due to the bioisosteric nature (e.g. Purines [3, 4] and plant alkaloids [5, 6]) and a wide range of activities. For example, the azolo[1,2,4]triazine derivatives including nucleotide analogs Riamilovir and Remdesivir (Fig. 1) exhibit antiviral properties [7, 8].

Characteristic examples of halogenated pyrazole and triazine-based drugs
The changes in the structural motifs of a molecule are primarily responsible for the non-covalent interactions with the active sites [9]. Therefore, an understanding of such relationships is critical for the developing of novel biologically potent compounds [10,11,12]. Recently, we have explored pyrazolo- [13,14,15,16,17] and pyrrolo[1,2,4]triazines with a moderate antimicrobial activity [18]. In the present work, the structures and packing modes, as well as non-covalent interactions in a series of functionalized pyrazolo[5,1-c][1,2,4]triazines were analyzed by X-ray single crystal diffraction and the Hirshfeld surface analysis.
Experimental
Synthesis of Compounds
8-Cyano-(1a), 8-carbethoxy-(1b), 8-nitro-(1c), 8-bromo-(1d)-7-bromo-3-tert-butylpyrazolo[5,1-c][1,2,4]triazin-4(1H)-ones were synthesized from the available 3-amino-5-tert-butyl-2-methylsulfanyl-1,2,4-triazin-5(4H)-one in several steps, in accordance with the published methods [13, 14]. Alkylation of compounds 1a–d with tert-butyl bromoacetate or n-butyl bromide in the presence of triethylamine furnished the corresponding N(1)-substituted derivatives 2a–d [15] (Scheme 1). Reduction of the heterocyclic carbonyl in 2b,d gave 4-hydroxypyrazolotriazines 3a,b [16]. 3-tert-Butyl-4,8-dialkyl-1,4-dihydropyrazolo[5,1-c][1,2,4]triazines 5a,b were obtained via nucleophilic additions of Grignard reagents to the triazine ring in aromatic precursors 4a,b (Scheme 2) [17]. 7-Bromo-3-tert-butyl-4-butyl-8-methylpyrazolo[5,1-c][1,2,4]triazine 6 was isolated in a good yield by treatment of 5b with N-bromosuccinimide and triethylamine [17].

Synthesis of compounds 2a–d and 3a,b

Synthesis of compounds 5a,b and 6
X-ray quality crystals of DMSO solvated 1a–c and unsolvated 2a,c, 3a,b, 5a,b and 6 were obtained by slow evaporation of solvents from the concentrated solutions in EtOAc/DMSO (10:1 v/v, for 1a–c and 6) or EtOAc/n-hexane (5:1–2:1 v/v, for other compounds) at r.t.
X‑Ray Crystal Structure Determination
X-ray diffraction data were collected at 100 K on a Bruker Quest D8 diffractometer equipped with a Photon-III area-detector (graphite monochromator, shutterless φ- and ω-scan technique), using Mo Kα-radiation (0.71073 Å). The intensity data were integrated by the SAINT program [19] and were corrected for absorption and decay using SADABS [20]. The structures were solved by direct methods using SHELXT [21] and refined on F2 using SHELXL-2018 [22]. All non-hydrogen atoms were refined with individual anisotropic displacement parameters. Locations of atoms H1A, H1B and H1C (1a), H1 (1c, 3a, 5a,b) were found from the electron density-difference map; they were refined with individual isotropic displacement parameters. All other hydrogen atoms were placed in ideal calculated positions and refined as riding atoms with relative isotropic displacement parameters. Aspherical scattering factors [23] were applied to all atoms in 1b excluding Br at the final steps of the refinement. The use of the aspherical model reduces the R-factor, which significantly improves the quality of crystal processing. Compound 6 is refined as an inversion twin, Flack parameter is 0.029(11). A rotating group model was applied for methyl groups in 6.
The SHELXTL [19] and Olex2 [24] program suites were used for molecular graphics. Displacement ellipsoids are set to the 50% probability level on all figures below. Crystal data, data collection and structure refinement details for 1a–c·DMSO, 2a,c, 3a,b, 5a,b and 6 are summarized in Tables 1 and 2. Crystal data for compounds 2a (CCDC 2017996) and 2c (CCDC 2017997) have been previously described in literature [15]. The structures 1a–c·DMSO, 3a,b, 5a,b and 6 have been deposited at the Cambridge Crystallographic Data Center with the reference CCDC numbers 2077359, 2077358, 2077349, 2077361–2077365; they also contain the supplementary crystallographic data. These data can be obtained free of charge from the CCDC via http://www.ccdc.cam.ac.uk/data_request/cif.
Results and Discussion
Crystal Structure Descriptions
Compounds 1a,c crystallize in the monoclinic crystal systems as 1:1 solvates with DMSO [1a]·DMSO, [1c]·DMSO. Crystals of compound 1b (triclinic) also obtained as a solvate of the composition [1b]3·DMSO. Other analyzed heterocycles 2a,c, 3a,b, 5a,b and 6 crystallize without inclusion of any solvent molecules in the crystal lattice.
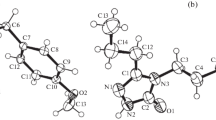
7-Bromo-3-tert-butylpyrazolo[5,1-c][1,2,4]triazin-4(1H)-ones 1a–c and 2a,c feature fully conjugated heterocyclic systems with a negligible deviation from the planarity. The nitrile, ester and nitro group were also significantly conjugated with the bicycle, which leads to the shortening of the C8–Cexocycl. bonds in all compounds (Table 3) and stabilization of the coplanar configuration between C–O or N–O and C7–C8 in 1b,c, 2c (Figs. 2, 3). Reduction of the C4 carbonyl group in 2b,d gave heterocyclic hemiaminals 3a,b and consequently led to change of the triazine conformation from planar to boat (Fig. 4, Table 3). This process also ruined the conjugation throughout the bicycle, which is substantiated by the elongation of the C3–C4 and N1–N2 bonds (from 1.47–1.48 and 1.35–1.36 Å in 2a,c to 1.52–1.53 and 1.38 Å in 3a,b, respectively). Compound 3b exhibit disorder of the N5–C4H–OH fragment in a single crystal, the minor component possessed cis-orientation of the OH and Boc groups.

The molecular structure of 1a,c·DMSO. H-atoms of alkyl groups are omitted

The molecular structure of [1b]3·DMSO. H-atoms of alkyl groups are omitted

The molecular structure of 3a,b. H-atoms of alkyl groups in 3a are omitted
The molecular structures of oxygen-free heterocycles 5a,b and 6 are shown in Fig. 5 and Table 4. The pyrazole cycle is nearly planar in all compounds, while the triazine cycle in 1,4-dihydro derivatives 5a and 5b showed boat conformations. The butyl and propyl groups in 5a,b are axial. It is interesting to note that the deviation from the plane is unequal, due to the C3 and C4 atoms with substituents in neighboring positions. For example, the torsion angles C8a–N5–C4–C3 and N5–C8a–N1–N2 are − 29.4(3)°, 21.9(3)° respectively for compound 5b, and 40.6(3)°, − 28.6(4)° respectively for 5a.

The molecular structure of 5a,b and 6. H-atoms of alkyl groups are omitted
On the contrary, the C(14) atom of the butyl group in aromatic pyrazolo[5,1-c][1,2,4]triazine 6 is located in the plane of the triazine cycle (the maximum deviation is ~ 5°). The N1–N2, N2–C3 and C3–C4 bond lengths changed from 1.399(2), 1.285(2) and 1.517(3) Å (5b) to 1.315(3), 1.380(4) and 1.374(4) (6), which indicated the significant conjugation. The lengths of analogous bonds in molecules 5a and 5b are similar with a deviation of 0.01–0.02 Å.
Intermolecular Interactions
Among the analyzed compounds, only 1b and 1c possessed intramolecular hydrogen bonds of the types C7C=O⋯H–N1, C7C–(Et)O⋯H–N1 (1b) and C7N–O⋯H–N1 (1c). The crystal structures of the 1a–c DMSO solvates showed intermolecular hydrogen bonds Me2S–O⋯H–N1, the experimental XRD O⋯N lengths lied within 2.6479(9)–2.7153(11) Å. In the case of [1b]3·DMSO, the crystals also contained symmetrical H-bonds of the type C7C=O⋯H–N1 for the neighboring molecules (O⋯N = 2.8655(11)–2.9591(11) Å, Figs. 3, 6).

Packing of compounds 1b,c
Alkylation of the N1 position in 1a,b to give the corresponding products 2a,c completely eliminated any hydrogen bonds in the crystals. On switching from C4=O to C4–OH, the resulting molecules of 3a,b appear to form hydrogen-bonded dimers via C4–OH⋯N6 with the O⋯N distances of 2.7972(12)–2.836(3) Å (Fig. 7). For 3b, an additional C4–OH⋯O=COt-Bu intermolecular bond was observed for the minor component (Table 5). The most significant short contacts between the halogens [25] within the series were observed for 1a (Br⋯Br = 3.8270(4) Å). Distinct halogen bonds with the Br⋯Br distances of 3.4782(3) Å and ∠C7–Br⋯Br = 141.65(3) (C7–Br⋯Br–C7, θ = 180.00(7)°) were observed only for 3a. The notable π–π stacking interactions within the series of 4-oxo derivatives were observed only for 2c and 3b with the minimum distances between the rings of 3.266 Å (2c), 4.814 Å (3b). The packing of the molecules is represented by endless pillars of stacked dimers arranged along the a (2c) or b (3b) axes.

Packing of compounds 3a, 5b, 6
In the crystals of 5a and 5b (Fig. 5, Table 4), molecules are stabilized by intermolecular hydrogen bonds of the type N1–H…N6 (2.03(4) Å, 163(3)° and 2.17(2) Å, 165(2)° respectively). In the crystal structure of aromatic compound 6 (Figs. 5, 7), π–π interactions with a distance of 3.693 Å were observed (the centroids are calculated as N5/N6/C7–C8a rings). The short contacts between the C7 and C8 atoms (3.394 Å) also worth mentioning. Compound 5b exhibited distinct halogen bonds with the Br⋯Br distances of 3.5513(4) Å (∠C7–Br⋯Br = 136.71(6)°; C7–Br⋯Br–C7, θ = 180.0(1)°, Fig. 7). However, on switching to aromatic triazine analog 6 the interhalogen distance increased to 3.8677(4) Å, while angle ∠C7–Br⋯Br was 79.38(8)° (Fig. 7). This is probably due to the increased contribution of π-stacking between the planar heterocyclic rings in 6 to the energy of intermolecular interactions.
Hirshfeld Surface Analysis
The Hirshfeld surface analysis [26] was carried out using the CrystalExplorer21 [27] to visualize and study the intermolecular contacts. The crystallographic information files (CIF) of compounds 1a,c, 2a, 3a,b, 5a,b and 6 were used as input for the analysis.
The intermolecular energies in crystal packing and the fingerprint plots with dnorm surfaces were calculated at B3LYP/6-31G(d,p) level of theory for all compounds. These contacts are reflected on the graph of dnorm = di + de surfaces as red, blue and white sections for dnorm respectively as shorter, longer and equal to the sums of Van der Waals radii. The dnorm surfaces and the fingerprint plots for compounds 1a,c, 2a, 3a,b, 5b and 6 are shown in Figs. 8, 9. The highest contributions to the Hirshfeld surfaces were made by the H⋯H contacts for all of the studied compounds. Analysis of the surfaces also revealed the presence of the halogen-heteroatom Br⋯X (X = N, O, Figs. 8, 9) type of short contacts for compounds 1a (C8–C≡N⋯Br), 1c (C8–N–O⋯Br), and 2a (C4=O⋯Br), which lengths were in excellent agreement with the experimental. Bromine-rich heterocycle 3b did not exhibit any significant Br⋯Br halogen bonds in the crystal, however, both analogs 3a and 3b showed HBu⋯Br contacts with the calculated lengths of 3.058–3.390 Å (Fig. 8).

The dnorm surfaces for 1a,c, 2a, 3a,b and their overall 2D fingerprint plots (bottom). Blue and red regions are weak and strong interactions, respectively (Color figure online)

The dnorm surfaces and energy frameworks for compounds 5b and 6
For compounds 5a,b, the significant contributions to the energy of non-valent interactions were made by the N⋯H bonds. On the contrary, hydrogen bonds were not observed in the crystal structure of compound 6, which corresponded to the XRD data (Fig. 7). The energy frameworks calculations were also performed for compounds 5b and 6 (Fig. 9). The obtained data indicate the pairwise intermolecular interaction energies in the crystal structures, as the radii of the visualized cylinders are proportional to the strength of these interactions. The strongest interactions were observed between the aromatic rings in 6, which confirms the presence of π⋯π stacking in the crystal structure.
Conclusion
To summarize, a total of eight novel azolo[1,2,4]triazine derivatives including 7-bromo-3-tert-butylpyrazolo[5,1-c][1,2,4]triazin-4(1H)-ones, 1,4-dihydropyrazolo[5,1-c][1,2,4]triazin-4-ols, 1,4-dihydro- and aromatic 3-tert-butyl-8-methylpyrazolo[5,1-c][1,2,4]triazines were investigated by X-ray diffraction analysis. The data obtained for the analyzed DMSO solvates and non-solvated crystals of compounds indicated the presence of intra- as well as inter-molecular short contacts of different nature and composition. The structural preferences and various packing modes based on the intermolecular interactions including H-bonds of the types O⋯H, N⋯H, as well as the π-stacking and halogen bonds were analyzed by the Hirshfeld surface and energy framework analysis. The presence of polar groups in the analyzed structures led to an increase in the number of intermolecular interactions, provided that there are no steric hindrances. The reduction of the triazine carbonyl led to change of the molecular conformation from planar to boat, ruined the conjugation and gave hydrogen-bonded dimers in the single crystals. On the other hand, aromatization of the bicyclic system completely eliminated any H-bonds, and significantly increased the contribution of π-stacking. The halogen-heteroatom and halogen-hydrogen types of short contacts dominated over the expected halogen bonds within the series.
Supplementary Material
CCDC 2077359, 2077358, 2077349, 2077361–2077365 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures.
Data Availability
The datasets generated and analysed during the current study are available from the corresponding author on request.
References
Maji M, Panja D, Borthakur I, Kundu S (2021) Org Chem Front 8:2673–2709
Kerru N, Gummidi L, Maddila S, Gangu KK, Jonnalagadda SB (2020) Molecules 25:1909
Cao T, Martini ML, Park KS, Kaniskan HÜ, Jin J (2022) 8.02—pyrimidines and their benzo derivatives, vol 8. Elsevier, Oxford, pp 86–228
Sharma V, Chitranshi N, Agarwal AK (2014) Int J Med Chem 2014:202784
Debnatha B, Singh WS, Das M, Goswami S, Singh MK, Maiti D, Manna K (2018) Mater Today Chem 9:56–72
Ain Q-U, Khan H, Mubarak MS, Pervaiz A (2016) Front Pharmacol 7:292
Wu X, Yu K, Wang Y, Xu W, Ma H, Hou Y, Li Y, Cai B, Zhu L, Zhang M, Hu X, Gao J, Wang Y, Qin H, Zhao M, Zhang Y, Li K, Du Z, Yang B (2020) Engineering 6:1199–1204
Eastman RT, Roth JS, Brimacombe KR, Simeonov A, Shen M, Patnaik S, Hall MD (2020) ACS Cent Sci 6:672–683
Guryanov I, Fiorucci S, Tennikova T (2016) Mater Sci Eng C 68:890–903
Dao P, Lietha D, Etheve-Quelquejeu M, Garbay C, Chen H (2017) Bioorg Med Chem Lett 27:1727–1730
Kumar A, Singh UK, Gupta P, Muzaffar F, Pathak P, Tomar PK (2016) Pharma Chem 8:259–273
Sherin DR, Geethu CK, Prabhakaran J, Mann JJ, Kumar JSD, Manojkumar TK (2019) Comput Biol Chem 78:108–115
Ivanov SM, Mironovich LM, Rodinovskaya LA, Shestopalov AM (2017) Russ Chem Bull 66:727–731
Ivanov SM (2021) Russ J Org Chem 57:151–159
Ivanov SM (2020) Tetrahedron Lett 61:152404
Ivanov SM, Koltun DS (2022) Tetrahedron Lett, submitted
Ivanov SM, Dmitrienko AO, Medvedev MG, Mironovich LM (2019) J Organomet Chem 896:168–182
Ivanov SM, Tuzharov EI, Kolotyrkina NG (2021) Russ J Gen Chem 91:2453–2461
Bruker, (2018) APEX-III. Bruker AXS Inc., Madison, Wisconsin, USA
Krause L, Herbst-Irmer R, Sheldrick GM, Stalke D (2015) J Appl Cryst 48:3–10
Sheldrick GM (2015) Acta Cryst A 71:3–8
Sheldrick GM (2015) Acta Cryst C 71:3–8
Lübben J, Wandtke CM, Hübschle CB, Ruf M, Sheldrick GM, Dittrich B (2019) Acta Cryst A 75:50–62
Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H (2009) J Appl Cryst 42:339–341
Cavallo G, Metrangolo P, Milani R, Pilati T, Priimagi A, Resnati G, Terraneo G (2016) Chem Rev 116:2478–2601
Spackman MA, McKinnon JJ (2002) CrystEngComm 4:378–392
Spackman PR, Turner MJ, McKinnon JJ, Wolff SK, Grimwood DJ, Jayatilaka D, Spackman MA (2021) J Appl Cryst 54:1006–1011
Author information
Authors and Affiliations
Contributions
The authors SMI and DSK contributed equally to the current manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ivanov, S.M., Koltun, D.S. Crystal Structure Analysis of 4-Oxo, 4-hydroxy- and 4-alkyl-7-bromopyrazolo[5,1-c][1,2,4]triazines. J Chem Crystallogr 53, 345–356 (2023). https://doi.org/10.1007/s10870-022-00973-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10870-022-00973-x