
Abstract
Deletions of chromosome 17p, where TP53 gene locates, are the most frequent chromosome alterations in human cancers and associated with poor outcomes in patients. Our previous work suggested that there were p53–independent mechanisms involved in chromosome 17p deletions-driven cancers. Here, we report that altered arachidonate metabolism, due to the deficiency of mouse Alox8 on chromosome 11B3 (homologous to human ALOX15B on chromosome 17p), contributes to the B cell malignancy. While the metabolites produced from lipoxygenase pathway reduced, chromosome 11B3 deletions or Alox8 loss, lead to upregulating its paralleling cyclooxygenase pathway, indicated by the increased levels of oncometabolite prostaglandin E2. Ectopic PGE2 prevented the apoptosis and differentiation of pre-B cells. Further studies revealed that Alox8 deficiency dramatically and specifically induced Cox-2(Ptgs2) gene expression. Repressing Cox-2 by its shRNAs impaired the tumorigenesis driven by Alox8 loss. And, in turn, tumor cells with Alox8 or 11B3 loss were sensitive to the COX-2 inhibitor celecoxib. This correlation between COX-2 upregulation and chromosome 17p deletions was consistent in human B-cell lymphomas. Hence, our studies reveal that the arachidonate metabolism abnormality with unbalanced ALOX and COX pathways underlies human cancers with 17p deletions and suggest new susceptibility for this disease.
Similar content being viewed by others


Introduction
Chromosome 17p is the most frequent deleted chromosome region in various human cancers, and is often associated with drug resistance and short survival [1, 2]. In lymphoid malignancies, chromosome 17p deletions (del(17p)) occur in 10-20% non-Hodgkin’s lymphoma like diffused large B-cell lymphoma (DLBCL) or mantle lymphoma, and 5-10% chronic lymphocytic leukemia [1, 3, 4]. Although del(17p) has been observed and widely used as a diagnostic marker in human cancers decades ago, its functional roles and potential underlying mechanisms are still unclear. With a conditional knockout mouse model of chromosome 11B3, the syntenic region in mice to human chromosome 17p13.1 and containing >100 coding genes, we showed that del(17p), as a whole, could promote tumorigenesis in multiple tumor models more than p53 loss itself [2]. These results suggested that besides TP53, additional tumor suppressor genes (TSGs) would be in this region.
Recently, our work and others’ have shown that PHF23, EIF5A, KCTD11, MAP2K4 and HIC1 would be putative TSGs on chromosome 17p [5,6,7,8]. To better understand the tumor suppression mechanisms of chromosome 17p, we performed an in vivo tumorigenesis screening with an shRNA library against the 99 coding genes on chromosome 11B3 in mice. Multiple potential TSGs were scored in this screening, including the plant homeodomain finger protein 23 (PHF23) and arachidonate 15-lipoxygenase type B (ALOX15B) [2, 8]. PHF23 is a reader for histone 3 lysine 4 tri-methylation and negatively regulates the deacetylase activity of HDAC through a new epigenetic regulatory complex, the PSH complex. Thus, the PSH complex coordinates two active histone markers, H3K4me3 and H3K27ac, for gene activation. Loss of PHF23 represses the expressions of differentiation-related genes and other downstream TSGs and thus promotes tumorigenesis [8]. Besides this PHF23-regulated epigenetic mechanism, metabolic alterations have been observed in cancers with del(17p). ALOX15B deficiency leads to accumulation of its substrate, arachidonic acid (AA), and accelerates Myc-driven lymphoma. Interestingly, these 17p TSGs collaborate with p53 to inhibit tumorigenesis, which might contribute to the super tumor suppression capacity of chromosome 17p as a whole [9]. However, the metabolic mechanism in cancer cells with del(17p) or ALOX15B loss has not been well understood. ALOX15B is a member of iron-containing polyunsaturated fatty acid metabolism enzymes, which oxygenate AA and generate a series of products, including hydroperoxy eicosatetraenoic acids (HpETEs), hydroxyeicosatetraenoic acid (HETEs) and leukotrienes. AA is a polyunsaturated fatty acid and abundant in many types of cells. AA can be metabolized through three distinct pathways, the cyclooxygenase pathway, the lipoxygenase pathway and the cytochrome P450 pathway, into numerous biologically active metabolites. In humans, there are six lipoxygenases and interestingly, five of them, ALOX15B, ALOX12B, ALOXE3, ALOX12 and ALOX15, reside in chromosome 17p. AA metabolism has been implicated in multiple biological processes and human diseases, such as inflammation, myocardial infarction, cholesterol metabolism, and cancer [10,11,12]. ALOX15B and ALOX12 in lymphoma have been reported to have a tumor-suppressive role in promoting murine lymphomas, while ALOX5, the only one not on chromosome 17p, seems to be required at least for some types of leukemia [2, 11, 13]. In contrast, cyclooxygenases, especially COX-2, and their products, such as prostaglandin E2 (PGE2), seem to have oncogenic roles in various types of cancers [14, 15]. Given these paradoxical functions of AA metabolism, it would be interesting to investigate how AA metabolism is disturbed in del(17p) and ALOX15B deficient tumors and their potential functions in tumorigenesis and treatment.
In this study, we profiled the metabolites derived from AA in del(17p) and ALOX15B deficient lymphoma cells by liquid chromatography-mass spectrometry (LC-MS). We analyzed the in vitro and in vivo functions of the cyclooxygenase pathway in these tumors with genetic and pharmaceutic assays. We found that upregulating the cyclooxygenase pathway contributed to del(17p) cancers’ biology and implied new vulnerability for this disease.
Results
Prostaglandin E2 is specifically enriched in del(17p) and Alox8-deficient cells
Our previous work has shown that arachidonic acid (AA) was accumulated in murine B-cell lymphomas with Alox8-knockdown or chromosome 11B3 deletion, which is syntenic to human chromosome 17p13 [2]. Here, we collected peripheral white blood cells from B-cell chronic lymphocytic leukemia (B-CLL) patients at West China Hospital, and measured AA amount through liquid chromatography-mass spectrometry (LC-MS). We found that cellular AA levels were significantly higher in del(17p) CLL (n = 3) than no del(17p) ones (n = 5) (Fig. 1A). Given that AA can be oxygenated into diverse bioactive lipid metabolites, we wondered how these elevated AA levels would alter the AA-derived metabolites in del(17p) tumors. Therefore, we profiled the AA metabolites, produced through either lipoxygenase or cyclooxygenase pathway, in primary mouse B-cell lymphomas with or without 11B3 loss by LC-MS. As Fig. 1B shown, two AA derivatives oxygenated by the cyclooxygenase pathway, prostaglandin E2 (PGE2) and thromboxane B2 (TXB2), were increased in 11B3-deleted lymphoma cells, while multiple metabolites produced from arachidonate lipoxygenases, including hydroxyeicosatetraenoates (HETEs) were reduced in 11B3-deleted cells compared to Trp53 loss only lymphoma cells. Interestingly, the levels of several other ALOX products, such as several hydroxy docosahexaenoic acids (HDoHEs) derived from docosahexaenoic acid (DHA) were decreased. Altogether, the AA metabolism is largely disrupted in del(17p) tumor cells, with a downregulated lipoxygenase pathway and upregulated cyclooxygenase pathway.

A AA levels in fresh patients’ white blood cells enriched with chromosome 17p-deleted CLL cells, compared to those without 17p deletion, measured by liquid chromatography-mass spectrometry (LC-MS). n = 3 for no del(17p) group and n = 5 for del(17p) group. Error bar represents “Mean with SEM”, *p < 0.05 (unpaired two-tailed t-test). B Profiling AA metabolites in mouse primary lymphoma cells with chromosome 11B3 deletion (11B3+/-), compared to Trp53-/- lymphoma cells, by LC-MS (n = 3). C AA levels in shAlox8 vs shRen Ba/F3 cells, measured by LC-MS and ELISA, respectively. n = 4 for shRen group and n = 8 for shAlox8 group. Error bar represents “Mean with SEM”, *p < 0.05 (unpaired two-tailed t-test). D AA levels in Alox8+/+ vs Alox8-/- Ba/F3 cells measured by LC-MS and ELISA, respectively. n = 6 for Alox8+/+ group and n = 4 for Alox8-/- group. Error bar represents “Mean with SEM”, ***p < 0.001 (unpaired two-tailed t-test). E Profiling AA metabolites in primary shAlox8 lymphoma cells, compared to shTrp53 lymphoma cells, by LC-MS (n = 3). F PGE2 levels in Myc; shTrp53-shRen (shRen) or Myc; shTrp53-shAlox8 (shAlox8) lymphoma cells, measured by ELISA. n = 4 for shRen group and n = 6 for shAlox8 group. Error bar represents “Mean with SEM”, *p < 0.05 (unpaired two-tailed t-test).
Given that there are five ALOX genes on chromosome 17p, next we tested whether ALOX15B loss would be able to mimic del(17p) and directly led to the AA metabolism dysregulation. In a previous study, we showed that AA was increased in mouse NIH3T3 fibroblast cells with Alox8 knockdown, homologous to human ALOX15B [2]. Here, we confirmed this observation in Ba/F3 cells, a murine pro-B cell line that is more relative to B cell lymphoma. Ba/F3 cells infected with shAlox8 had significantly higher levels of AA than that in cells with shRen, measured by LC-MS (Fig. 1C). Similar results of higher AA levels were observed in the corresponding medium of Ba/F3 cells with shAlox8 (Supplementary Fig. 1A). Further, we generated Ba/F3 cells with Alox8 loss-of-function mutations by CRISPR/Cas9 technology, verified by T7 endonuclease I assay and Sanger sequencing (Supplementary Fig. 1C and D). The cellular and medium AA levels were significantly higher in Alox8-mutated cells (Alox8-/-) than in control cells (Fig. 1D and Supplementary Fig. 1B), consistently with what we observed in shAlox8 cells. Then we profiled the AA metabolites by LC-MS in primary B-cell lymphoma cells with or without shAlox8. In consistent with the finding in del(11B3) lymphoma cells, multiple AA downstream metabolites produced from cyclooxygenase pathway, such as PGE2, PGD2 (Prostaglandin D2) and TXB2, were upregulated in Alox8-deficient lymphoma cells (shAlox8;Myc), compared with control lymphoma cells (shTrp53;Myc) (Fig. 1E). The accumulation of PGE2 in tumor cells with Alox8 loss was further confirmed by enzyme-linked immunosorbent assay (ELISA) (Fig. 1F). These data suggested that Alox8 loss contributed to the enrichment of AA and it derives like PGE2 via cyclooxygenase pathway in tumor cells with del(17p).
RNA-seq analyses reveal a transcriptomic similarity between Alox8-deficient cells and those treated with PGE2
To decipher the molecular consequences of Alox8 loss, we analyzed the transcriptomes of pre-B cells with Alox8 or control Renilla shRNAs by RNA sequencing (RNA-seq). Both the unsupervised hierarchical clustering and principal component analysis (PCA) plot showed that the RNA-seq results of pre-B cells with shAlox8 were grouped together, distinguished from shRen cells (Fig. 2A and Supplementary Fig. S2A). Gene ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways enrichment analysis show that 154 GO terms and 26 KEGG pathways were significantly downregulated in shAlox8 pre-B cells, compared to control shRen cells. Among them were immune response-related pathways such as cytokine-cytokine receptor interaction, chemokine signaling pathway, cellular response to interleukine-1, response to lipopolysaccharide, and the apoptosis pathway significantly downregulated in shAlox8 cells (Fig. 2B and Supplementary Fig. S2B). Gene set enrichment analysis (GSEA) results revealed that the upregulated genes by PGE2 treatment were positively enriched in shAlox8 pre-B cells, compared to shRen cells (NES = 1.135). In contrast, the downregulated genes by PGE2 treatment were negatively enriched (NES = −1.119) (Fig. 2C) [16]. The transcriptomic similarity further confirmed the altered cyclooxygenase pathway by Alox8 loss.

A Unsupervised clustering of RNA-seq data of shAlox8 or shRen pre-B cells. n = 4, B KEGG enrichment analysis of genes downregulated in shAlox8 cells comparing to shRen cells. C–E Gene set enrichment analysis of genes belonging to the top 100 differentially expressed genes with PGE2 treatment (C), HALLMARK_P53_PATHWAY and HALLMARK_APOPTOSIS gene sets (D) or GO_LYMPHOCYTE_DIFFERENTIATION and GO_B_CELL_DIFFERENTIATION gene sets (E) in shAlox8 cells comparing to shRen cells. NES, normalized enrichment score; FDR, false discovery rate. F The percentage of Annexin V + 7-AAD- apoptotic cells of pre-B cells treated with different concentrations of PGE2, measured by flow cytometry. n = 3. *p < 0.05 (unpaired two-tailed t-test). G, H The percentage of IgM+ in B220 + B cells as measured by flow cytometry analysis. n = 3. Error bar represents “Mean with SEM”, **p < 0.01(unpaired two-tailed t-test).
PGE2 increase prevents cellular apoptosis and B-cell differentiation
Gene set enrichment analysis (GSEA) results also showed that HALLMARK_P53 PATHWAY (NES = −1.546, FDR q = 0.013) and HALLMARK_APOPTOSIS pathway gene sets (NES = −1.560, FDR q = 0.011) were significantly negatively enriched in the transcriptome of shAlox8 cells compared to that of shRen cells (Fig. 2D). Multiple lymphocyte differentiation and B cell differentiation gene signatures were negatively enriched in Alox8-deficient pre-B cells (Fig. 2E). The enrichment of these pathways might underlie the anti-apoptosis and differentiation block effects of Alox8 deficiency.
To test whether upregulated PGE2 by Alox8 loss contributed to apoptosis and B-cell differentiation, we treated pre-B cells, the assumable cell-of-origin for Myc-driven B lymphoma in our model, with PGE2. The results showed that ectopic PGE2 prevented the apoptosis of pre-B cells, measured by Annexin V and 7-AAD staining, in a dosage-dependent manner (Fig. 2F). Further, we found that the differentiation of pre-B cells, indicated by IgM expression, was significantly repressed by PGE2 treatment (Fig. 2G, H). The survival advantage and differentiation block suggested that PGE2 might promote the transformation of B progenitor cells.
Cox-2 (Ptgs2) but not Cox-1 (Ptgs1) is induced by Alox8 deficiency
Cyclooxygenase is the key enzyme to catalyze AA into prostaglandins, including PGE2. There are two cyclooxygenases COX-1 and COX-2, encoded by PTGS1 and PTGS2, respectively. It is known that while PTGS1 is constitutively-expressed, PTGS2 gene transcription could be rapidly induced in response to stimuli [17]. Consistent with the increased levels of the cyclooxygenase pathway metabolites, we found that Ptgs2 but not Ptgs1 gene expression was dramatically induced up to 250-fold in Alox8-deficient lymphoma cells, compared to that in control shRen lymphoma cells (Fig. 3A). The synergistically increased expressions of PTGS2, PTGES and its metabolite PGE2 indicated a shift of the AA metabolism from the lipoxygenase pathway to the cyclooxygenase pathway.

A Relative mRNA levels of Ptgs1(Cox-1) and Ptgs2(Cox-2) in shRen or shAlox8 lymphoma cells, measured by RT-qPCR. n = 3 for shRen group and n = 6 for shAlox8 group. *p < 0.05 (unpaired two-tailed t-test). B Knockdown efficiency of Ptgs2 by shPtgs2.562 and shPtgs2.3383 in shTrp53 lymphoma cells, measured by RT-qPCR. n = 3. Error bar represents “Mean with SEM”, *p < 0.05 (unpaired two-tailed t-test). C Kaplan–Meier tumor-free survival of recipient mice transplanted with tandem shRNAs for both Alox8 and Ptgs2. n = 6. *p < 0.05 (log-rank test). D Relative cell counts of Myc; shTrp53-shRen (shRen) or Myc; shTrp53-shAlox8 (shAlox8) lymphoma cells treated with 50 μM celecoxib or vehicle for 48 h. n = 3 for shRen group and n = 5 for shAlox8 group. Error bar represents “Mean with SEM”, *p < 0.05 (unpaired two-tailed t-test). E Relative cell counts of Trp53-/- or 11B3+/- lymphoma cells treated with 50μM celecoxib or vehicle for 48 h. n = 3 for shRen group and n = 4 for shAlox8 group.
Upregulation of cyclooxygenase pathway is required for Alox8 loss-driven lymphomagenesis
Because of the disrupted AA metabolism balance between the lipoxygenase pathway and the cyclooxygenase pathway in Alox8-deficient tumors and the known oncogenic functions of PGE2 in various cancers, we hypothesized that the enhanced cyclooxygenase pathway might mediate the tumor-promoting function of Alox8 loss. Two independent shRNA against Ptgs2 were cloned and validated by qRT-PCR (Fig. 3B). Ptgs2 shRNAs were cloned into a retrovirus-based vector carrying Myc cDNA and tandem shRNAs (shAlox8.2865-shPtgs2.562 or shAlox8.2865-shPtgs2.3383), with which Alox8 and Ptgs2 would be simultaneously repressed in the same cell with Myc overexpression. These tandem shRNA constructs were introduced into B progenitor pre-B cells, followed by transplantation into sub-lethally irradiated recipient C57BL/6 mice. The recipient mice were monitored daily by palpation, and the tumor-free survivals were recorded. Kaplan–Meier curve showed that recipient mice transplanted with shAlox8.2865-shRen transduced pre-B cells developed lymphomas with a median latency of 17 days, similar to those with shAlox8.2865 only [2]. However, compared to control shAlox8-shRen recipient mice, mice carrying shAlox8-shPtgs2 pre-B cells developed lymphomas with significantly longer tumor latencies (Fig. 3C; Median onset 27 and 39 days, respectively). Intriguingly, although the resulting lymphoma cells containing shAlox8-shPtgs2 as enriched GFP + cells in enlarged lymph nodes, these cells have upregulated Ptgs2 gene expression to a similar level as that in control shAlox8-shRen lymphoma cells (data not shown) and displayed similar histological features (Supplementary Fig. 3A, B), suggesting that shPtgs2 suppressive effect on Ptgs2 mRNA level had been bypassed or compensated during lymphomagenesis. Altogether, these results strongly indicated that the cyclooxygenase pathway upregulation, following Alox8 loss, was required for lymphomagenesis driven by Alox8 deficiency.
The AA metabolism alteration with the lipoxygenase-cyclooxygenase pathway unbalances implied that a gain of vulnerability for cancers with del(17p) or ALOX15B loss. As a proof-of-concept, we treated lymphoma cells with shRen or shAlox8 with celecoxib, a commonly used anti-inflammation drug inhibiting COX-2. The results showed that shAlox8 tumor cells were significantly more sensitive to COX-2 repression than the control cells (Fig. 3D). Further, we tested the inhibitory effect of celecoxib on mouse del(11B3) lymphoma cells. Tumor cells with del(11B3) also displayed a better response to celecoxib treatment than those with only p53 loss (Fig. 3E). The susceptibility of these Alox8 deficient tumor cells to celecoxib suggested that targeting the unbalanced AA metabolism might be a new treatment for cancers with del(17p) in patients.
The unbalanced lipoxygenase pathway and cyclooxygenase pathway in lymphoma patients with del(17p) suggest new susceptibility
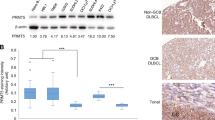
Given that there are multiple tumor suppressor genes like p53 and PHF23 on chromosome 17p, we wondered how much ALOX15B repression and the resulted aberrant AA metabolism would contribute to del(17p) cancer. GSEA results showed that the genes upregulated in shAlox8 cells were significantly positively enriched in human chromosome 17p-deleted DLBCL cells (NES = 1.74, FDR q = 0.03). And in contrast, the genes downregulated in shAlox8 cells were significantly negatively enriched in del(17p) DLBCL (NES = −1.48, FDR q = 0.03) (Fig. 4A). These results suggest that Alox8 loss partially mimicked the molecular characteristics of del(17p). The cyclooxygenase pathway was upregulated in both Alox8 deficient and 11B3-deleted mouse lymphoma cells, indicated by increased levels of Ptgs2 expression and its product PGE2 (Figs. 1E and 1F). Here, we further tested the expressions of PTGS2 in human del(17p) tumors. Since the expression levels of genes on chromosome 17p are reduced in 17p-deleted tumors, we used the corrected expression levels of chromosome 17p13 genes to indicate 17p deletions. By analyzing the transcriptomes of 48 DLBCL samples in the TCGA cohort, we found that the expression levels of PTGS2 were significantly negatively correlated with those of chromosome 17p13 genes (Pearson = −0.46, p < 0.01) (Fig. 4B). A similar correlation was also observed in the TCGA-AML cohort (Pearson = −0.33, p < 0.001) (Fig. 4C). The negative correlation between 17p genes and PTGS2 was consistent with our hypothesis that the cyclooxygenase pathway of AA metabolism was upregulated in del(17p) cancers.

A GSEA plots showing that genes were chosen from top 200 upregulated (shAlox8_UP) or downregulated (shAlox8_DOWN) genes in shAlox8 cells comparing to shRen cells significantly positively or negatively enriched in human chromosome 17p-deleted DLBCL cells. B, C Correlations between expression levels of PTGS2 and averages of chromosome 17p genes in TCGA-DLBCL or TCGA-AML. Data analyzed from TCGA-DLBCL (n = 48) or TCGA-AML (n = 187).
Taken together, our studies revealed the AA metabolism alterations in del(17p) cancers. The deficiency of the co-deleted 17p TSG ALOX15B led to a repressed lipoxygenase pathway and an enhanced cyclooxygenase pathway for AA metabolism (Fig. 5). Moreover, this metabolism abnormality implied a new susceptibility to this recalcitrant malignancy.

Schematic representation showing that arachidonate lipoxygenase and cyclooxygenase pathway in normal cell (left) or lymphoma cells with chromosome 17p deletions (right).
Discussion
Metabolism reprogramming has been shown to be a hallmark of cancer as early as a century ago when Warburg’s effect was first described [18, 19]. Besides numerous studies on glucose metabolism, there are emerging evidences showing that altered lipid metabolism has significant effects on tumorigenesis not only through providing essential cellular components but also as signaling molecules [20,21,22]. AA, a polyunsaturated fatty acid with a 20-carbon chain, can be converted into multiple bioactive lipids through three distinct metabolism pathway, the lipoxygenase pathway, the cyclooxygenases pathway and the cytochrome P450 pathway. Some of these AA-derived products have been shown to be involved in inflammation, cancer and other human diseases [23]. In this study, we have illustrated that cyclooxygenases/prostaglandins, upregulated by lipoxygenase ALOX15B loss, contributed to the tumor formation and maintenance of Alox8-deficient lymphoma. COX-2 is known to be generally absent in most normal cells but is highly enriched in many malignant cells like colorectal, lung, breast and prostate cancers; and PGE2, the major bioactive metabolite of COX genes, can promote the proliferation of cancer cells, enhance their stemness and protect them from apoptosis [24]. Furthermore, the high expression of PTGS2, encoding COX-2, is also correlated with poor prognosis in lymphomas [25]. In contrast, ALOX15B is recognized to have an anti-carcinogenic effect as its downregulated expression in various cancers and its loss driving lymphoma development [2, 26,27,28]. Our work revealed that these two enzymes can crosstalk. Loss of Alox8 specifically induced Ptgs2 expression, inhibiting which would prevent Alox8-deficient B progenitor cells transformed into lymphoma, and suppress Alox8-deficient lymphoma cell growth. PTGS2 is suggested to be induced in response to various cellular and environmental stresses. There are some signal pathways like NF-κB and PI3K/AKT pathway reported to regulate PTGS2 gene transcription [29, 30]. We found that Alox8 loss activated the PI3K-mTOR pathway (data not shown). But, whether the upregulated PI3K-mTOR pathway is downregulated Alox8 cell induces PTGS2 gene expression needs future study.
Chromosome 17p deletions are one of the most frequently deleted chromosome regions that happened in almost all known cancers. Our previous work and other laboratory studies have demonstrated that chromosome 17p deletions have an impact on cancer biology beyond TP53 loss. Multiple tumor suppressor genes like PHF23, EIF5A, KCTD11, MAP2K4 and ALOX15B, have been reported to promote tumorigenesis when lost, indicating that gene expression regulation, signal transduction, arachidonate lipoxygenase and other cellular pathways are altered with the loss of chromosome 17p [2, 5, 6, 8]. Here, our results indicate that the arachidonate cyclooxygenase pathway also plays a role in 17p-deleted tumor biology. We showed that ALOX15B deficiency with chromosome 17p deletions was demonstrated to promote lymphomagenesis and maintain corresponding lymphoma cell growth through upregulating COX-2 level, as discussed before. Moreover, high expression of COX-1 and especially COX-2 are correlated with chromosome 17p loss. Loss of mouse chromosome 11B3, homologous to human chromosome 17p13, has a high level of the product of the cyclooxygenase pathway, PGE2. More importantly, 11B3-deleted lymphoma cells were more sensitive to COX-2 inhibitor celecoxib. These results support that arachidonate cyclooxygenase/prostaglandins contribute to the chromosome 17p-deleted cancer biology. How these various dysregulated pathways in chromosome 17p deletions cooperatively work during the progression of malignancy and response to commercial therapies are under study.
Besides ALOX15B, there are four other arachidonate lipoxygenases locating on chromosome 17p, including ALOX12, ALOX12B, ALOX15 and ALOXE3. Among them, Alox12 was reported to be required for the tumor suppression functions of p53 in an Eµ-Myc mouse lymphoma model [13]. Consistently, we found knockdown Alox12 could drive Myc-overexpressed pre-B cells transformed into lymphoma in recipient mice (data not shown). These tumor-suppressive ALOX genes are clustered in chromosome 17p suggested that their loss in 17p-deleted cancers may have a cooperative impact on tumor development. Conversely, the only ALOX family gene outside of chromosome 17p, ALOX5 on chromosome 10, did not have the same tumor-suppressing role in our mouse lymphoma model. Instead, other reports show that ALOX5 may have opposite roles in a different context that it promoted BCR-ABL driven B-cell acute lymphoblastic leukemia while inhibited MLL-fusion driven acute myeloid leukemia [11, 31]. Interestingly, ALOX5 product 5-HETE was significantly increased in mouse chromosome 11B3-deleted lymphoma cells compared to that in Trp53-/- control. Inhibition of ALOX5 has a synthetic effect with COX-2 inhibitor on suppressing cancer cell growth. Therefore, it may be a promising therapeutic strategy to treat 17p-deleted lymphomas with both ALOX5 and COX-2 inhibitors.
Materials and methods
Mice
All mice experiments were approved by the Institutional Animal Care and Use Committees of Sichuan University. Mouse lymphomagenesis experiments were performed as previously described [2]. Briefly, B220 + B progenitor cells isolated from bone marrow were transduced with retroviruses followed by tail-vein injection into sublethally irradiated C57BL/6 mice. Lymphomas were monitored weekly with disease state being defined by palpable enlarged solid lymph nodes. Tumor measurements were performed blindly. All recipient mice were divided into each group randomly before transplantation. Tumor measurements were performed blindly.
Human samples
Primary peripheral blood cells from patients with CLL were collected at the outpatient department of West China Hospital during 2017–2018. The status of chromosome 17 was confirmed by fluorescence in situ hybridization (FISH). A total of 10 patients were recruited, including 6 with 17p deletion, and 4 with intact 17p. Peripheral blood samples were transported at 4 °C and lysed using ACK lysing buffer within 24 h. After lysing, 2 × 106 white blood cells were prepared for intracellular lipids extraction. The Ethics Committee approved the study of West China Hospital of Sichuan University [HX-2019-(114)].
Cell line culture
Ba/F3 (#HB-283), 293 T (#CRL-1573) were from ATCC. Ba/F3 cells were maintained in RPMI 1640 medium supplemented with 10% v/v FBS and 2 ng/mL IL-3(cat. No.:CP39; Novoprotein). 293 T cells were maintained in DMEM medium supplemented with 10% v/v FBS. All cell lines were routinely tested for Mycoplasma by PCR. Experiments were performed within 4 weeks after fresh viable cells were thawed.
Retroviral construction
shRNAs were cloned into MSCV-mirE-SV40-Myc-IRES-GFP retroviral construct. Retrovirus packaging and infection of cells were done as previously reported [2].
ELISAs
Prostaglandin E2 (PGE2) was measured using an ELISA kit (cayman, CAS 363-24-6) according to the manufacturer’s instructions. Arachidonic acid (AA) was measured using an ELISA kit (Elabscience, E-EL-0051c) according to the manufacturer’s instructions.
LC-MS
Cells were extracted with chloroform/methanol and washed with 0.9% saline. The lipid-containing chloroform phase was obtained. Standard curves were constructed by least-squares linear regression analysis using the peak area ratio of a given eicosanoid over its reference IS against the calibrator’s nominal concentration. UPLC-MS/MS analyses were conducted on an Agilent UPLC-MS/MS system consisting of 1290 UPLC-system coupled with an Agilent 6470 triple-quadrupole mass spectrometer (Agilent Technologies, USA). Peak determination and peak area integration was performed with MassHunter Workstation software (Agilent, Version B.08.00), while auto-integration was manually inspected and corrected if necessary. Appropriate internal standards corrected the obtained peak areas of targets (IS) and calculated response ratios were used throughout the analysis. All experiments were repeated more than twice.
RNA-sequencing analysis
RNA-seq were sequenced by BGISEQ500 sequencing machine with 50-bp single-end reads. The RNA-seq reads were aligned to the reference genome (GRCm38) by STAR_2.6.0a. Transcript abundance was normalized and measured in Transcripts Per Kilobase Million (TPM). Differential gene expressions were analyzed by DESeq2. Genes with absolute fold changes >1 and FDR ≤ 0.05 were counted as differentially expressed genes. Samples distance were calculated by PCA and the Euclidean distance. GSEA was applied to show statistically significant similarities and differences between two given clusters by identifying a priori-defined gene set. Our model was ordered to obtain a rank-ordered gene list and the top 100 high and low expressed genes in the gene set GSE1195214 and GSE209445 as grp files. Followed by running GSEA, TCGA DLBCL patients split into 17 P deletion and no 17 P deletion, 17 P deletion VS no 17 P deletion Fold change to obtain a rank-ordered gene list, and the top 200 high and low expressed genes in shAlox8. Followed by running GSEA.
All the transcriptome data in the TCGA database were transformed by log2(X + 1). Correlations between PTGS2 expression levels and the mean expression levels of 97 genes in 17p detected in TCGA-LAML and TCGA-DLBC were calculated by Pearson’s correlation coefficient and statistic powers were quantified followed by a two-sided hypothesis test and 0.95 confidence level.
Quantitative PCR
RNA was isolated with Trizol, cDNA was synthesized with QuantStudio™ 3 Real-Time PCR System (Thermo Fisher Scientific). All experiments were repeated more than twice. A list of all primers used for PCR analysis is given in Supplementary Table 1.
In vitro drug response assays
Lymphoma cell lines generated from shTrp53-shRen, shTrp53-shAlox8.1252, shTrp53-shAlox8.2865 tumor-bearing mice were cultured in BCM medium (45% DMEM, 45% IMDM, 10% FBS, 2 mM glutamine, 50 μM β- mercaptoethanol, 1× penicillin/streptomycin) in 96-well plates. Cells were treated with the indicated concentrations of celecoxib (Selleck; S1261) for 2 days. The celecoxib dissolved in DMSO. The number of living cells was determined by BD Accuri C6. All experiments were repeated more than twice.
Statistical analysis
Statistical analysis was performed by GraphPad Prism9 (RRID: SCR_002798). Kaplan–Meier tumor-free survival were analyzed by log-rank test. Other statistical significance was examined by unpaired two-tailed t-test.
Data availability
All data generated or analyzed during this study are included.
References
Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343:1910–6.
Liu Y, Chen C, Xu Z, Scuoppo C, Rillahan CD, Gao J, et al. Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature. 2016;531:471–5.
Bea S, Colomo L, Lopez-Guillermo A, Salaverria I, Puig X, Pinyol M, et al. Clinicopathologic significance and prognostic value of chromosomal imbalances in diffuse large B-cell lymphomas. J Clin Oncol. 2004;22:3498–506.
Bea S, Ribas M, Hernandez JM, Bosch F, Pinyol M, Hernandez L, et al. Increased number of chromosomal imbalances and high-level DNA amplifications in mantle cell lymphoma are associated with blastoid variants. Blood. 1999;93:4365–74.
Scuoppo C, Miething C, Lindqvist L, Reyes J, Ruse C, Appelmann I, et al. A tumour suppressor network relying on the polyamine-hypusine axis. Nature. 2012;487:244–8.
Ahn YH, Yang Y, Gibbons DL, Creighton CJ, Yang F, Wistuba II, et al. Map2k4 functions as a tumor suppressor in lung adenocarcinoma and inhibits tumor cell invasion by decreasing peroxisome proliferator-activated receptor gamma2 expression. Mol Cell Biol. 2011;31:4270–85.
Wales MM, Biel MA, Deiry WE, Nelkin BD, Issa J, Cavenee WK, et al. p53 activates expression of HIC-1, a new candidate tumour suppressor gene on 17p13.3. Nat Med. 1995;1:570–7.
Chen M, Chen X, Li S, Pan X, Gong Y, Zheng J, et al. An epigenetic mechanism underlying chromosome 17p deletion–driven tumorigenesis. Cancer Discov. 2021;11:194–207.
Chen M, Yang Y, Liu Y, Chen C. The role of chromosome deletions in human cancers. Adv Exp Med Biol. 2018;1044:135–48.
Demetz E, Schroll A, Auer K, Heim C, Patsch JR, Eller P, et al. The arachidonic acid metabolome serves as a conserved regulator of cholesterol metabolism. Cell Metab. 2014;20:787–98.
Chen Y, Hu Y, Zhang H, Peng C, Li S. Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat Genet. 2009;41:783–92.
Romano M, Cianci E, Simiele F, Recchiuti A. Lipoxins and aspirin-triggered lipoxins in resolution of inflammation. Eur J Pharm. 2015;760:49–63.
Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. 2019;21:579–91.
Gupta RA, Dubois RN. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer. 2001;1:11–21.
Wun T, McKnight H, Tuscano JM. Increased cyclooxygenase-2 (COX-2): a potential role in the pathogenesis of lymphoma. Leuk Res. 2004;28:179–90.
Sanin DE, Matsushita M, Klein Geltink RI, Grzes KM, van Teijlingen Bakker N, Corrado M, et al. Mitochondrial membrane potential regulates nuclear gene expression in macrophages exposed to prostaglandin E2. Immunity. 2018;49:1021–1033.e1026.
Kang YJ, Mbonye UR, DeLong CJ, Wada M, Smith WL. Regulation of intracellular cyclooxygenase levels by gene transcription and protein degradation. Prog Lipid Res. 2007;46:108–25.
Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8:519–30.
Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47.
Rohrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer. 2016;16:732–49.
Eberlin LS, Gabay M, Fan AC, Gouw AM, Tibshirani RJ, Felsher DW, et al. Alteration of the lipid profile in lymphomas induced by MYC overexpression. Proc Natl Acad Sci USA. 2014;111:10450–5.
Fishbein A, Wang W, Yang H, Yang J, Hallisey VM, Deng J, et al. Resolution of eicosanoid/cytokine storm prevents carcinogen and inflammation-initiated hepatocellular cancer progression. Proc Natl Acad Sci USA. 2020;117:21576–87.
Das UN. Arachidonic acid in health and disease with focus on hypertension and diabetes mellitus: A review. J Adv Res. 2018;11:43–55.
Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010;10:181–93.
Hazar B, Ergin M, Seyrek E, Erdogan S, Tuncer I, Hakverdi S. Cyclooxygenase-2 (Cox-2) expression in lymphomas. Leuk Lymphoma. 2004;45:1395–9.
Pidgeon GP, Lysaght J, Krishnamoorthy S, Reynolds JV, O’Byrne K, Nie D, et al. Lipoxygenase metabolism: roles in tumor progression and survival. Cancer Metastasis Rev. 2007;26:503–24.
Jiang WG, Watkins G, Douglas-Jones A, Mansel RE. Reduction of isoforms of 15-lipoxygenase (15-LOX)-1 and 15-LOX-2 in human breast cancer. Prostaglandins Leukot Essent Fatty Acids. 2006;74:235–45.
Suraneni MV, Moore JR, Zhang D, Badeaux M, Macaluso MD, DiGiovanni J, et al. Tumor-suppressive functions of 15-Lipoxygenase-2 and RB1CC1 in prostate cancer. Cell Cycle. 2014;13:1798–810.
Jang BC, Kim DH, Park JW, Kwon TK, Kim SP, Song DK, et al. Induction of cyclooxygenase-2 in macrophages by catalase: role of NF-kappaB and PI3K signaling pathways. Biochem Biophys Res Commun. 2004;316:398–406.
Jang BC, Paik JH, Kim SP, Shin DH, Song DK, Park JG, et al. Catalase induced expression of inflammatory mediators via activation of NF-kappaB, PI3K/AKT, p70S6K, and JNKs in BV2 microglia. Cell Signal. 2005;17:625–33.
Wang Y, Skibbe JR, Hu C, Dong L, Ferchen K, Su R, et al. ALOX5 exhibits anti-tumor and drug-sensitizing effects in MLL-rearranged leukemia. Sci Rep. 2017;7:1853.
Acknowledgements
We thank all the lab members in Liu and Chen’s laboratory for their invaluable advice and suggestions. We thank Dr. Yuquan Wei for generous support. We thank Dr. Huiru Tang (Fudan University) and Dr. Yulan Wang (Nanyang Technological University) for their technical support for LC-MS detecting AA metabolites. We thank the Core Facilities of West China Hospital for technical support. This work was supported by the National Natural Science Foundation of China (82130007, 81670182), the Sichuan Science and Technology Program (2018JZ0077, 2020YJ0381), 1.3.5. Project for Disciplines of Excellence, West China Hospital, Sichuan University (ZYJC21009, ZYGD22012), and Scientific Research Project of Nanchong Municipal Science and Technology Bureau, Sichuan Province (19SXHZ0443).
Author information
Authors and Affiliations
Contributions
ZX, LQ and YL designed the experiments. ZX, LQ, PL, MC, QZ, HH, MT and DW performed the experiments. YL, CC, LD, ZX and LQ analyzed data. XP and XC performed bioinformatics analyses. ZX, LQ, CC and YL wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qi, L., Pan, X., Chen, X. et al. COX-2/PGE2 upregulation contributes to the chromosome 17p-deleted lymphoma. Oncogenesis 12, 5 (2023). https://doi.org/10.1038/s41389-023-00451-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41389-023-00451-9
This article is cited by
-
Murine Alox8 versus the human ALOX15B ortholog: differences and similarities
Pflügers Archiv - European Journal of Physiology (2024)

{kind=link}
{kind=link}
{kind=link}