
Abstract
Cohen syndrome (CS) is a rare multisystem autosomal recessive disorder associated with mutations in VPS13B (vacuolar protein sorting homolog 13B). The NAPB-related neurodevelopmental disorder is characterized mainly by early-onset epileptic encephalopathy (EOEE) and is associated with mutations in NAPB that encodes for SNAP-beta (soluble NSF attachment protein beta). Here we describe male triplets, clinically presenting with the phenotype of subtle but distinctive facial features, intellectual disability, increased body weight, neonatal EOEE, and prominently variable abnormal behaviors of autism and sexual arousal. The EEG showed multifocal epilepsy, while the brain MRI showed no abnormalities. Diagnostic exome sequencing (ES), the applied next-generation sequencing approach, revealed the interesting finding of two novel homozygous variants in two genes: VPS13B missense variant (c.8516G > A) and NAPB splice-site loss (c.354 + 2 T > G). Sanger sequencing verified the segregation of the two recessive gene variants with the phenotype in family members. The prediction algorithms support the pathogenicity of these variants. Homozygosity mapping of ES data of this consanguineous family revealed multiple chromosomal regions of homozygosity stretches with the residing of VPS13B (chr8: 100830758G > A) and NAPB (Chr20: 23,375,774 A > C) variants within the largest homozygous blocks further supporting the disease-genes causal role. Interestingly, the functions of the two proteins; VPS13B, a transmembrane protein involved in intracellular protein transport, and SNAP-beta involved in neurotransmitters release at the neuronal synaptic complexes, have been associated with Golgi-mediated vesicular trafficking. Our ES findings provide new insights into the pathologic mechanism underlying the expansion of the neurodevelopmental spectrum in CS and further highlight the importance of Golgi and Golgi-membrane-related proteins in the development of neurodevelopmental syndromes associated with early-onset non-channelopathy epilepsy. To our knowledge, this is the first report documenting multifocal EOEE in CS patients with the association of a pathogenic NAPB variant.
Similar content being viewed by others

Introduction
Cohen syndrome (CS) (OMIM #216,550) is a rare autosomal recessive multisystem disorder, characterized by a spectrum of global developmental delay, intellectual disability, hypotonia, facial dysmorphism, ocular defects, neutropenia, endocrine and skeletal abnormalities [1, 2]. Associated seizures and behavioral disturbances were described in a few cases of CS, in which cortical brain malformation or hippocampus atrophy was demonstrated [3, 4]. Clinical heterogeneity in CS was evident among patients from different ethnic backgrounds [5], genetic isolates as well as patients of the same ancestry [6]. The syndrome has been described in diverse ethnicities, but in very few Arab families: Omani, Lebanese, and Tunisian [5, 7]. Investigators relate the heterogeneity in the appearance of certain clinical features involving short stature, delayed puberty, blindness on top of retinal dystrophy, or neutropenia to the ethnic background of the patients’ population [8]. Mutations in the vacuole protein sorting 13B (VPS13B) gene, also known as COH1 [OMIM *607817], are recognized to underlie the characteristic features in CS [5]. VPS13B protein, one of the four members of the human VPS13 protein family that share a common feature of being giant proteins > 300 KD in size, is a Golgi peripheral membrane protein that co-localizes with Golgi matrix proteins. Silencing of VPS13B in vitro and in CS patients’ fibroblasts revealed fragmentation and disorganization of Golgi structures, hence its suggested roles in maintaining Golgi integrity and intracellular trafficking of Golgi-derived vesicles [9].
On the other hand, mutations in the NAPB gene [N-ethylmaleimide-sensitive factor (NSF)-attachment protein beta, OMIM *611270] that encodes for the SNAP-beta, a protein component of the SNARE (SNAP-receptor) complex, have been described in association with early-onset epileptic encephalopathy (EOEE) characterizing the multifocal intractable seizures in disorders of neurodevelopmental nature [10,11,12,13]. SNAP-beta plays a basic role in the fusion/priming of neuronal synaptic vesicles and the release of neurotransmitters [10].
This study aims to find out the genetic defect underlying the unexplained phenotype of intellectual disability, mild dysmorphism, developmental delay, EOEE, and prominent behavioral patterns in male triplets. Herein, we report the interesting genetic findings identified in Palestinian male triplets, who were found to carry novel homozygous recessive variants in VPS13B and NAPB, and present with CS phenotype, EOEE, and variably prominent behavioral abnormalities.
Subjects and methods
Subjects
Six subjects, three male probands (single-placenta triplet) and three family members (mother, father, and a healthy sibling), were subjected to diagnostic exome sequencing genetic testing and analysis. The male triplets were on follow-up in the Neurology Clinic, Hamad Medical Corporation, Qatar, and first attended the Genetic Clinic, at the age of 23 years. Written informed consent was obtained from the participants and patients’ guardians for the data release, capturing photos, and publication.
Methods
In-house clinical investigations, complete blood pictures, EEG, brain imaging (MRI), and fragile-X testing (outsourced) were performed. Blood samples of the six family members were collected and sent to GeneDx for diagnostic exome sequencing (ES).
Exome and Sanger sequencing
Diagnostic exome sequencing (ES) was carried out at GeneDx, using genomic DNA from the probands, parent, and a healthy sibling. The exonic regions and flanking splice junctions of the genome were captured using the IDT xGen Exome Panel v1.0. Massively parallel (NextGen) sequencing was done on an Illumina system with 100-bp paired-end reads. Reads were aligned to human genome build GRCh37/UCSC hg19 and analyzed for sequence variants using a custom-developed analysis tool. Sanger sequencing was applied to verify the segregation of VPS13B and NAPB variants with the phenotype in all participants of the family members.
Prediction algorithms
VarSome,Footnote 1 the human genomic variant search engine [14], the Ensembl variant Effect PredictorFootnote 2 web tool, and GeneDx variants assertion criteria were applied for variants’ interpretation. Variants classified as pathogenic or variant of uncertain significance (VUS) according to the American College of Medical Genetics and Genomics (ACMG) guidelines [15].
Homozygosity mapping (HZM)
HZM is a powerful tool for the prioritization and identification of recessively inherited disease-causing genes; the tool provides further supporting evidence for the candidature of the two gene variants. BAM-sequencing files of the proband A, parent, and healthy sibling were combined in one VCF, uploaded, and run on the HomozygosityMapper online tool. Gene Distiller functionality help to allocate the genes in the query (VPS13B on chromosome 8 and NAPB on chromosome 20) along the homozygous regions.Footnote 3 HZM aims to promote the genetic diagnosis’s reliability by looking at the homozygous stretches on the two chromosomes of interest, Chr8 and Chr20, and whether the corresponding gene was allocated within such homozygous blocks.
Results
Clinical presentation
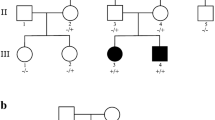
A triplet of male probands was born preterm at 36 weeks gestational age by caesarian section. The triplets had a single placenta and were blood group O + . The first-cousin parent has five other older normal offspring (Fig. 1a). The neonatal history described generalized hypotonia, limited spontaneous movements, but no feeding difficulties. Facial characteristics involved notably large, long, and protruding ears, bulbous nasal tip, and triangular face. Motor developmental delay was marked; head support was achieved at the age of 10 months, sat unsupported at 15 months, and walked at two and half years old. Psychomotor retardation and learning disabilities were evident but variable across the three probands (Table 1). At the first genetic assessment (age of 23 years), the probands displayed a facial gestalt of a long face, brushy and heavy eyebrows, long eyelashes, unilateral ptosis of the left eyelid, open mouth with the thick lower lip, short philtrum, short neck, long but not-protruding ears, bulbous nasal tip, and prominent nose (Fig. 1b). All probands showed joint hyperextensibility, bilateral big toes, wide space between the first and second toes, and cylindrical fingers. A history of being overweight as an adolescent was reported. The features of the male gender, long face, cognitive impairment, and psychomotor delay suggested fragile X syndrome as the first provisional clinical diagnosis; however, FMR1-CGG repeats testing proved normal.

Family pedigree and patients’ photos. a First cousin consanguineous Palestinian family, filled rectangular denotes affected male triplets, empty circles and boxes denote healthy subjects. b Facial appearance of the male triplets at the age of 23 years shows long face, heavy eyebrows, prominent nose and bulbous nasal tip, short philtrum, open mouth with thick lower lip, long ears, and short neck
Epilepsy and behavioral disturbances
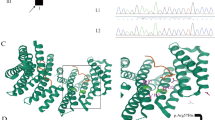
Epilepsy was a prominent neonatal feature that variably progressed in the triplet. In proband A, the seizures occurred usually during sleep; the patient would awaken looking scared or in discomfort, may scream with loss of awareness, stiffen diffusely, and may exhibit a head turn and bilateral jerking movements. His seizures were frequent, occurring daily, sometimes in clusters with the attacks typically lasting for a minute. The patient is currently on lamotrigine, levetiracetam, and an escalating dose of clobazam with only partial improvement. He failed many other trials of anti-seizure drugs, and specifically developed symptomatic hyperammonemia on valproate. The VEEG (video electroencephalogram) data with interictal and ictal recordings support multifocal epilepsy (Fig. 2A). Despite the protracted nature of the seizures, his brain magnetic resonance imaging was normal (Fig. 2B). Proband B seizures were less frequent, occurring during wakefulness and sleep, and fit the description for classical generalized tonic–clonic (GTC) seizures that last for a few seconds. A more subtle seizure type was sometimes noted with a sudden slowing in speech fluency, echolalia, and blank stare for a minute or less. At the age of 16 years, a vagal nerve stimulator device (VNS) was implanted. At 23 years of age, the VNS was turned off because of suspected worsening sleep apnea. His seizures came under good control on valproic acid after it was added to lamotrigine and clonazepam. A childhood EEG recorded the presence of multifocal epileptiform discharges. Proband C had the mildest presentation of epilepsy; his seizures were well controlled by a combination of antiepileptic medication (Table 1) and became quite infrequent.

Brain MRI, EEG-recording, and chest X-ray. a) Proband A, interictal EEG samples in a referential montage showing multifocal independent sharp waves and spikes: 1, left temporal. 2, right temporal. 3, left frontal. 4, right frontal. b) Proband B, axial T1 (1) and T2 (2) brain MR images at the level of the basal ganglia showing no significant abnormality. Sagittal FLAIR (3) and coronal T2 (4) images showing normal size of the corpus callosum and comparable temporal lobes especially the hippocampal regions with no signal abnormality. c) and d) Plain PA-radiography of the chest; c proband A, blunting of the right costophrenic angle (thick arrow) with clear left one. Bilateral bony cervical ribs are seen longer on the right side (blue thin arrows), d proband B, small right sided bony cervical rib (thin arrow), left sided subcutaneous VNS battery noted
Behavioral abnormalities
Behavioral abnormalities were particularly significant in proband B, who has the full clinical picture of autism, as well as exaggerated sexual arousal, and maladaptive behaviors. Proband C showed a much milder spectrum of autism, while proband A displayed no recognizable features of autism.
Skeletal abnormalities
The cervical ribs, seen in the available radiographs of probands A and B, is a newly reported radiological finding in CS (Fig. 2C and D). Spinal deformities or clubfoot was not identified in any of the triplets.
Molecular genetic findings
ES results validated by Sanger sequencing revealed two homozygous novel variants: a missense variant of uncertain significance, c.8516G > A, p.(Arg2839Gln) in exon 46 (NM_017890.4) of VPS13B and a splice-site loss pathogenic variant c.354 + 2 T > G in exon 4 (NM_001283018.1) of NAPB. The triplets were homozygous for both variants in VPS13B and NAPB, while the parent was heterozygous for these variants. The healthy sibling was a carrier for the NAPB variant but did not carry the VPS13B variant (Fig. 3A).

Sanger sequencing and HZM. a) Sanger validation and segregation of VPS13B and NAPB variants. Sanger sequencing showing the recessive co-segregation of the two variants in VPS13B and NAPB genes with the phenotype. b) Homozygosity mapping output of the combined ES-VCF. Upper panel: Demonstrating the extensive homozygous stretches on most of the chromosome. Lower panel: The genomic position of the homozygous stretches on chromosomes 8 and 20, the arrows point to the NAPB (Chr20: 23,375,774 A > C) and VPS13B (100830758G > A) gene variants residing within the homozygous stretches on chromosomes 20 and 8, respectively
In silicopredictive tools’ findings
The VPS13B missense variant is a strong Cohen-causing candidate: variant pathogenicity showed as damaging/pathogenic/or disease-causing in 12 prediction algorithms and as tolerated in only seven. The ACMG classified this variant as VUS based on the pathogenicity score, multiple computational tools that support a deleterious effect on the gene, and the very rare allele frequency in gnomAD exomes (= 0.0000159). The present missense VPS13B variant affects the same codon (c.8516) that has been previously altered by a nonsense variant (p.R2839X) in two independently reported CS patients [16]. The arginine, a polar-basic residue at the protein position P.2839, is highly conserved among species (phyloP100way = 9.6) and is replaced here by glutamine, a polar but neutral amino acid. The variant supporting criteria involve being observed in a homozygous status in the affected triplets, the phenotypic fit (the gene is highly specific for the disease presentation), and a high CADD score of 33. Our variant was reported on the CliniVar submission page by another independent clinical testing submitter.
NAPB (NM_001283018.1): c.354 + 2 T > G, a splice site loss variant, is a strong candidate for the NAPB-related neurodevelopmental disorder. Regarding variant pathogenicity, it has been shown as damaging/pathogenic/or disease-causing in seven prediction tools and has not been reported as tolerated in any computational prediction tools. The clinical significance of this variant was interpreted as “pathogenic” based on the criteria of being a “null” variant (very strong), not found in any available population databases, and has a strongly conserved position (phyloP100way = 9.09). This variant has the supportive criteria of being homozygous in the triplets and the gene is highly linked to EOEE neurodevelopmental disorder. The NAPB variant decreases the splicing efficiency by 80% as predicted by MaxEntScan; there is no detectable nearby alternative splice site that could result in exon skipping. This variant has a CADD score of 34.
The two gene variants co-segregate in a recessive pattern with the family phenotype (Fig. 3A).
Homozygosity mapping
ES-BAM files, uploaded on the HomozygosityMapper online tool revealed extensive homozygous stretches on most of the chromosomes. VPS13B variant (chr8: 100830758G > A) and NAPB variant (Chr20: 23,375,774 A > C) were residing within one of the large homozygous blocks identified on chromosomes 8 and 20, respectively (Fig. 3B).
Discussion
This report is the first to describe Cohen syndrome (CS) in probands of Arab-Palestinian origin. Clinical heterogeneity in CS presentation is well-acknowledged [5, 6]; however, CS has been reported in very few families of Arab ethnicity [7, 17]. Global developmental delay, learning disabilities, normal brain imaging, and distinctive facial gestalt were featured in the present triplets, in whom the CS diagnosis was first uncovered when they were 23 years old following a diagnostic ES. Features of neonatal/infant feeding difficulties, postnatal microcephaly, short stature, delayed puberty, myopia, retinal dystrophy, and neutropenia were absent in our patients. Earlier studies described neutropenia as an exclusive Caucasian feature and suggested a normal neutrophil count in CS patients of non-European descent [16]. The absence of neutropenia seemed to be a feature in Arab CS patients [16, 7, this report]. In terms of intellectual abilities, our triplets demonstrated variable degrees of intellectual impairment; while proband C had a profound intellectual disability, probands A and B exhibited moderate learning disabilities. This supports previous studies involving patients from different descends [18, 19].
The unique clinical observations in our patients involved the remarkable variability in the abnormal behavioral patterns and the early-onset (neonatal) protracted epilepsy. Autistic behavior in CS was acknowledged in some studies [6, 18, 20], while described as a rare event or non-observed in others [5]. Variability in the autistic association was demonstrated among unrelated patients of different series; however, in the present study, the variability was observed among three triplets that had a single placenta and were of the same gender and blood group suggesting the likelihood of being monozygotic. Proband C showed the full picture of autism in addition to significant sexual arousal and aggressive behaviors. Proband B had a mild spectrum of autism, while proband A showed no clinical features suggestive of autism. The partial penetrance of VPS13B homozygous mutation has been hypothesized to be a reason for the variability in autism association and cognitive involvements [21]. Herein, we suggest an alternative hypothesis that points to the extent of injury and fragmentation of Golgi structures, in the tissues including the brain, as an important contributor to the differential functional impact and variability in neurodevelopmental findings in CS patients. This assumption is supported by the knowledge that the Golgi peripheral membrane proteins contribute to Golgi integrity and proper orientation and consequently its normal subcellular function in protein and lipid post-translational modification. In addition, the glycosylation defects in CS have been presumed to be due to Golgi disorganization secondary to alteration in the transmembrane VPS13B protein [22]. The hypothetical role of VPS13B in Golgi-mediated vesicle trafficking and endolysosomal transport has been recently verified in VPS13BΔEx3/ΔEx3 mice model [23]. This model demonstrated the occurrence of Golgi apparatus mislocalization and its aberrantly configured membrane stacks. The authors provided experimental evidence for the role of VPS13B in the transport of Golgi-mediated vesicles during the formation of spermatid’s acrosome.
The exciting molecular result, in this report, is the sequencing finding of the NAPB splice-site pathogenic variant, which recessively segregates with the phenotype in family members. NAPB-related neurodevelopmental disorder (NAPB-related NDD) is characterized mainly by EOEE and seems of ultra-rare occurrence; to date, it was documented in only four independent families and in association with loss of function mutations, two nonsense mutations [11, 12], and a recent splice-site mutation [13]. Our report is the 5th family worldwide.
There is a clinical overlap between NAPB- and VPS13B-associated neurodevelopmental features of intellectual disability, developmental delay, and autistic spectrum; however, the EOEE characterizes the NAPB phenotype.
Epilepsy has been described as an uncommon feature and is, generally, poorly characterized within CS patients. In the present report, epilepsy was significantly prominent and of variable severity among the triplets. The three probands started the seizures at the age of 6 months; however, proband A has the most intractable and predominantly nocturnal epilepsy phenotype with the highest frequency of seizures. The epileptic events in proband B were equally distributed throughout wakefulness and sleep and of moderate severity. Proband C has the mildest course of epilepsy. A variable response to antiepileptic drugs defines an additional seizure-related variability. The seizures previously described in a few independent CS patients were characterized by an older age of onset between 18 and 24 months, benign courses, and being smoothly manageable by antiepileptic medications. The reported EEGs recorded a spike-wave epileptic discharge that tends to be unilateral, prevails over the temporal-parietal-occipital regions, and is continuous during slow waves of sleep (CSWS) [24]. In our patients, the EEG demonstrated multifocal discharges supporting a previously unrecognized CS-brain phenotype of multifocal neonatal-onset epilepsy. The causal role of NAPB’s mutations is supported by the documented association of altered SNAP-beta protein with early-onset multifocal epileptic encephalopathy (EOEE) in patients with unexplained neurodevelopmental disorders [11,12,13].
The present pathogenic NAPB variant expands the phenotypic spectrum of Cohen syndrome and likely explains a role in the associated prominent neurodevelopmental behavioral phenotype. The SNAP-beta has been recognized in the pathways (http://PathCards.genecards.org) involved in intra-Golgi traffic, Golgi-derived vesicle trafficking and transport, and Golgi-to-ER retrograde transport. In the same direction, VPS13B co-localizes with Golgi matrix proteins and demonstrated a role in Golgi integrity [9]. This pays the attention to the common role of VPS13B and SNAP-beta proteins in Golgi-medicated vesicular transport.
Conclusion
The family described here presents the first co-occurrence of CS and NAPB-related neurodevelopmental disorder in triplets from a consanguineous family. This report underscores the importance of studying the role of the Golgi membrane and functionally related proteins in the development of neurodevelopmental and behavioral disturbance disorders and adds to the ever-expanding landscape of genetic causes of early-onset syndromic non-channelopathy epilepsy.
Our family acknowledges the uniform finding of absent neutropenia in Arab CS patients, reported so far. Sexual arousal and aggressiveness that were significantly prominent and notably variable between the triplets expanded the clinical spectrum of the diseases.
Data availability
The exome sequencing data of this family is not publicly available. Inquiries to access the data can be made to the corresponding author.
References
Cohen M, Hall B, Smith D, Graham C, Lampert K (1973) A new syndrome with hypotonia, obesity, mental deficiency, and facial, oral, ocular and limb anomalies. J Pediatr 83:280–284. https://doi.org/10.1016/s0022-3476(73)80493-7
Kivitie-Kallio S, Norio R (2001) Cohen syndrome: essential features, natural history and heterogeneity. Am J Med Genet 102:125–136. https://doi.org/10.1002/1096-8628(20010801)102:2%3c125:aid-ajmg
Guerrini R, Genton P, Bureau M, Parmeggiani A, Salas-Puig X, Santucci M, Bonanni P et al (1998) Multilobar polymicrogyria, intractable drop attack seizures, and sleep-related electrical status epilepticus. Neurol 51:504–512. https://doi.org/10.1212/wnl.51.2.504
Atabek M, Keskin M, Kurtogelu S, Kumandas S (2004) Cohen syndrome with insulin resistance and seizure. Pediatr Neurol 30:61–63. https://doi.org/10.1016/s0887-8994(03)00309-6
Kolehmainen J, Black C, Saarinen A, Chandler K, Clayton-Smith J, Träskelin A, Perveen R et al (2003) Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle-mediated sorting and intracellular protein transport. Am J Hum Genet 72:1359–1369. https://doi.org/10.1086/375454
Douzgou S, Petersen M (2011) Clinical variability of genetic isolates of Cohen syndrome. Clin Genet 79:501–506. https://doi.org/10.1111/j.1399-0004.2011.01669.x
Rejeb I, Jilani H, Elaribi Y, Hizem S, Hila L, Zillahrdt J, Chelly J, Benjemaa L (2017) First case report of Cohen syndrome in the Tunisian population caused by VPS13B mutations. BMC Med Genet 18:134–139. https://doi.org/10.1186/s12881-017-0493-5
Falk M, Feiler H, Neilson D, Maxwell K, Lee J, Segall S et al (2004) Cohen syndrome in the Ohio Amish. Am J Med Genet 128A:23–28. https://doi.org/10.1002/ajmg.a.30033
Seifert W, Kühnisch J, Maritzen T, Horn D, Haucke V, Hennies H (2011) Cohen syndrome-associated protein, COH1, is a novel, giant Golgi matrix protein required for Golgi integrity. J Biol Chem 286:37665–37675. https://doi.org/10.1074/jbc.M111.267971
Burgalossi A, Jung S, Meyer G, Jockusch W, Jahn O, Taschenberger H, O’Connor V et al (2010) SNARE protein recycling by αSNAP and βSNAP supports synaptic vesicle priming. Neuron 68:473–487. https://doi.org/10.1016/j.neuron.2010.09.019
Conroy J, Allen N, Gorman K, Shahwan A, Ennis S, Lynch S, King M (2016) NAPB–a novel SNARE-associated protein for early-onset epileptic encephalopathy. Clin Genet 89:E1-3. https://doi.org/10.1111/cge.12648
Reuter M, Tawamie H, Buchert R, Gebril O, Froukh T, Thiel C, Uebe S et al (2017) Diagnostic yield and novel candidate genes by exome sequencing in 152 consanguineous families with neurodevelopmental disorders. JAMA Psychiat 74:293–299. https://doi.org/10.1001/jamapsychiatry.2016.3798
Zhao X, Wang Y, Cai A, Mei S, Liu N, Kong X (2021) A novel NAPB splicing mutation identified by trio-based exome sequencing is associated with early-onset epileptic encephalopathy. Eur J Med Genet 64:104101. https://doi.org/10.1016/j.ejmg.2020.104101
Kopanos C, Tsiolkas V, Kouris A, Chapple C, Aguilera M, Meyer R, Massouras A (2019) The human genomic variant search engine. Bioinforma 35:1978–1980. https://doi.org/10.1093/bioinformatics/bty897
Richard S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–423. https://doi.org/10.1038/gim.2015.30
Seifert W, Holder-Espinasse M, Kühnisch J, Kahrizi K, Tzschach A, Garshasbi M, Najmabadi H et al (2009) Expanded mutational spectrum in Cohen syndrome, tissue expression, and transcript variants of COH1. Hum Mutat 30:E404-420. https://doi.org/10.1002/humu.20886
Hennies H, Rauch A, Seifert W, Schumi C, Moser E, Al-Taji E, Tariverdian G et al (2004) Allelic heterogeneity in the COH1 gene explains clinical variability. Am J Hum Genet 75:138–145. https://doi.org/10.1086/422219
Chandler K, Moffett M, Clayton-Smith J, Baker G (2003) Neuropsychological assessment of a group of UK patients with Cohen syndrome. Neuropediatrics 34:7–133. https://doi.org/10.1055/s-2003-38617
Karpf J, Turk J, Howlin P (2004) Cognitive, language, and adaptive behavior profiles in individuals with a diagnosis of Cohen syndrome. Clin Genet 65:327–332. https://doi.org/10.1111/j.1399-0004.2004.00229.x
Howlin P, Karpf J, Turk J (2005) Behavioural characteristics and autistic features in individuals with Cohen syndrome. Eur Child Adolesc Psychiatry 14:57–64. https://doi.org/10.1007/s00787-005-0416-4
Yu T, Chahrour M, Coulter M, Jiralerspong S, Okamura-Ikeda K, Ataman B, Schmitz-Abe K et al (2013) Using whole-exome sequencing to identify inherited causes of autism. Neuron 77:259–273. https://doi.org/10.1016/j.neuron.2012.11.002
Duplomb L, Duvet S, Picot D, Jego G, Chehadeh-Djebbar S, Marle N, Gigot N et al (2014) Cohen syndrome is associated with major glycosylation defects. Hum Mol Genet 23:2391–2399. https://doi.org/10.1093/hmg/ddt630
Da Costa R, Bordessoules M, Guilleman M, Carmignac V, Lhussiez V, Courot H, Bataille A et al (2020) Vps13b is required for acrosome biogenesis through functions in Golgi dynamic and membrane trafficking. Cell Mol Life Sci 77:511–529. https://doi.org/10.1007/s00018-019-03192-4
Coppola G, Federico R, Epifanio G, Tagliente F, Bravaccio C (2003) Focal polymicrogyria, continuous spike-and-wave discharges during slow-wave sleep and Cohen syndrome: a case report. Brain Develop 25:446–449. https://doi.org/10.1016/s0387-7604(03)00055-x
Acknowledgements
The authors thank the patient’s family for approving the publication of the case. Open Access funding is provided by the Qatar National Library.
Funding
Open Access funding provided by the Qatar National Library.
Author information
Authors and Affiliations
Contributions
AA performed the clinical care genetic work-up, gene results’ interpretation and wrote the manuscript’s first draft. NH performed the neurological clinical care and EEG and contributed to their interpretation. GA assessed and revised the computational predictions of the variants. AC contributed to ES data analysis and interpretation (diagnostic ES at GeneDx). AE contributed to the radiographs interpretation. The authors revised the manuscript’s final version.
Corresponding author
Ethics declarations
Ethical approval
All participants gave written approval to participate in the study. No ethics committee approval was needed due to specific policies for case studies by Weill Cornell Medicine Qatar.
Consent for publication
The legal guardian of the triplet has signed an informed consent for capturing the photos and publishing their data and photos in scientific journal articles.
Conflict of interest
AC is an employee of GeneDx, Inc.; other authors declare no conflict of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
AbdelAleem, A., Haddad, N., Al-Ettribi, G. et al. Cohen syndrome and early-onset epileptic encephalopathy in male triplets: two disease-causing mutations in VPS13B and NAPB. Neurogenetics 24, 103–112 (2023). https://doi.org/10.1007/s10048-023-00710-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-023-00710-2