
Abstract
An accelerated de novo lipogenesis (DNL) flux is a common characteristic of cancer cells required to sustain a high proliferation rate. The DNL enzyme fatty acid synthase (FASN) is overexpressed in many cancers and is pivotal for the increased production of fatty acids. There is increasing evidences of the involvement of FASN in several hallmarks of cancer linked to its ability to promote cell proliferation via membranes biosynthesis. In this review we discuss about the implication of FASN in the resistance to cell death and in the deregulation of cellular energetics by increasing nucleic acids, protein and lipid synthesis. FASN also promotes cell proliferation, cell invasion, metastasis and angiogenesis by enabling the building of lipid rafts and consequently to the localization of oncogenic receptors such as HER2 and c-Met in membrane microdomains. Finally, FASN is involved in immune escape by repressing the activation of pro-inflammatory cells and promoting the recruitment of M2 macrophages and T regulatory cells in the tumor microenvironment. Here, we provide an overview of the involvement of the pro-oncogenic enzyme in the hallmarks of cancer making FASN a promising target in anti-cancer therapy to circumvent resistance to chemotherapies.
Similar content being viewed by others

A long enzyme in a few words
Fatty acid synthase (FASN), previously characterized as OA-519 for Oncogenic Antigen-519 [1], is a ubiquitous and cytosolic enzyme. FASN is the second enzyme of de novo lipogenesis (DNL) catalyzing the synthesis of palmitic acid (C16:0) by using malonyl-CoA and acetyl-CoA as substrates, and NADPH,H+, as co-substrate. FASN is functional as a head-to-tail [2] or head-to-head [3] homodimer. Each monomer exhibits seven distinct enzymatic activities working in a sequential and coordinated manner plus an acyl carrier protein (ACP) that carry the acyl group during its elongation [4]. The final product is released by hydrolysis catalyzed by the thioesterase (TE) domain of FASN. The complete structure of mammalian FASN has been solved by X-ray crystallography except for the flexible ACP and TE domains [5]. FASN is not only involved in energy storage in the form of triglycerides but it also actively participates in the synthesis of membrane components and second messengers, and, indirectly, in protein modification by acylation [4]. At the organism level FASN is essential for gestation [6], lactation [7], respiration [8] and ageing [9].
From a pathological point of view, the implication of FASN in tumorigenesis was first identified in human breast carcinoma cells in 1994 [10]. Thus, FASN is overexpressed in breast cancer [11] but also in many other types of cancers: colorectal [12], prostate [13], stomach [14], esophageal [15], lung [16], pancreatic [17], ovarian [18], hepatic [19, 20], melanoma [21], glioma [22] and in primary effusion lymphoma (PEL) [23]. FASN is often a poor prognostic marker, its overexpression being correlated to a decrease in the survival of patients. However, it is not considered as an oncogene as its overexpression in healthy cells does not induce a malignant transformation as shown for hepatocytes [24]. Through the years, many studies have highlighted the involvement of FASN in different hallmarks of cancer including cell metabolism, proliferation, migration, invasion, resistance to cell death, immune escape, and angiogenesis (Fig. 1): this makes the lipogenic enzyme an interesting and promising target in anti-cancer therapies.

The pro-oncogenic enzyme FASN is involved in sustained proliferation, dysregulation of cell energetics, resistance to cell death, induction of angiogenesis, activation of invasion and metastasis, and prevention of immune destruction. Adapted from Hanahan and Weinberg, 2011 [25].
FASN and the deregulation of cellular energetics
One of the hallmarks of cancer cells is metabolic reprogramming [25]. This includes an increased DNL unlike healthy cells which have a reduced lipogenesis, fatty acid needs being satisfied usually through dietary (exogenous) intake. The acceleration of lipogenesis observed in tumors promotes cell proliferation, survival, invasion and resistance to chemotherapy [26]. The increase in the lipogenic flux is partly related to the Warburg effect and to the overexpression of several metabolic enzymes including FASN. This increase in DNL flux has been not only described in hepatocellular carcinoma (HCC) [26] but also in breast cancer [27], glioblastoma [28], colorectal cancer [29] and in hematologic malignancies like acute myelogenous leukemia and endemic Burkitt lymphoma [30].
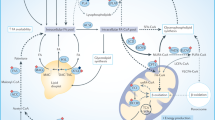
The highest levels of the lipogenic enzymes ATP citrate lyase (ACLY), acetyl-CoA carboxylase (ACC), stearoyl-coenzyme A desaturase 1 (SCD1) and FASN have been detected in HCC characterized by an aggressive phenotype [31, 32]. These enzymes are generally overexpressed in HCC explants in association with an increase of triglycerides, fatty acids and cholesterol compared to the adjacent non-tumoral tissue [31]. Recently, it has been shown in non-Hodgkin lymphoma that FASN promoted nucleotide biosynthesis required for supporting malignant proliferation [33]. By using NADPH,H+ to produce palmitate, FASN limits the level of the co-substrate and thus promotes 6-phosphogluconate dehydrogenase (PGDH) activity of which NADPH,H+ is an allosteric inhibitor. PGDH is the second enzyme of the pentose-phosphate pathway (oxidative branch) that generates ribulose-5-phosphate then converted into ribose-5-phosphate by the keto-isomerase (non-oxidative branch). As a result, FASN indirectly increases DNA/RNA synthesis required for cell proliferation. FASN also promotes protein synthesis through the activation of mammalian/mechanistic target of rapamycin (mTOR) activity in HepG2 [34] and in HCT116 cells [35] (Fig. 2).

Many enzymes are upregulated in cancer cells including the lipogenic enzymes ACLY, ACC, and FASN. Their overexpression leads to an increased lipid synthesis sustained by the Warburg effect. In addition FASN promotes mTOR activation resulting in an increased protein synthesis. FASN also favors the activity of the PPP (pentose-phosphate pathway) enzyme PGDH by increasing the pool of NADP+, a co-substrate of the latter. PPP upregulation concomitantly increases DNA/RNA synthesis. The dotted arrows represent an indirect effect. PGDH, 6-phosphogluconate dehydrogenase; AcCoA, acetyl-CoA; MalCoA, malonyl-CoA; TCA, tricarboxylic acid cycle; OAA, oxaloacetic acid; ACLY, ATP citrate lyase; ACC, acetyl-CoA carboxylase; PPP, pentose-phosphate pathway; mTOR, mammalian/mechanistic target of rapamycin; FASN, fatty acid synthase.
FASN and proliferative signaling
Overexpression of FASN in breast cancer cells promotes their survival and proliferation via the hyperactivation of HER1/2 receptors [11]. In turn, HER1/2 induce expression of FASN via activation of the MAPK and PI3K/Akt pathways (Fig. 3) highlighting a positive feedback to maintain high levels of FASN in cancer cells [11]. Jin et al. have demonstrated that inhibition of FASN by C75 (Table 1) in the breast cancer cell lines SKBR3 and BT474 was responsible for an increase in the internalization and degradation of HER2 associated with a decrease in the level of HER2 mRNA [36]. FASN promotes the expression and activation of HER2 probably by allowing its localization in lipid rafts and, conversely, HER2 favors the expression and activity of FASN (Fig. 3). The overexpression of FASN in prostate cancer cells (LNCaP) increases cell proliferation and soft agar growth [13]. In addition, mice bearing xenografts of esophageal squamous cell carcinoma cells (Colo680N) treated with the FASN inhibitor C93 (Table 1) showed a significant inhibition of tumor growth [15]. Together, these observations strongly support a role for FASN in cell proliferation and tumor growth.

FASN participates in the elaboration of lipid rafts required for signaling-associated receptors including the oncogenic receptors HER2 and EGFR. Thus, a positive feedback is ON as these receptors drive FASN expression via the activation of the MAPK and PI3K/Akt/mTOR pathways. PI3K and mTOR govern the expression of the transcription factor SREBP-1 which in turn induces FASN expression. mTOR also regulates FASN mRNA translation. In addition, FASN favors indirectly EGFR palmitoylation which prevents its inhibition by TKI. Finally, FASN promotes stemness and therefore cell proliferation.
FASN is overexpressed in breast cancer stem cells (BCSC), promotes stemness, and is hyperactivated in stemness-enriched samples [37]. Its inhibition with the antioxidant epigallocatechin-3-gallate (EGCG) or with the EGCG synthetic derivative G28 (Table 1) diminishes stemness and cell proliferation. It has been recently demonstrated that mTOR promotes FASN expression in HCC cells [34] and in breast cancer cells [38] (Fig. 3). In non-small cell lung cancer (NSCLC) cells with a mutant EGFR, FASN induces indirectly EGFR palmitoylation which prevents its inhibition by tyrosine kinase inhibitors (TKI). This results in a persistent signaling driven by mutated EGFR and consequently to cell proliferation [39] (Fig. 3). DNL is more active in glioblastoma CSCs compared to non-stem cancer cells [40]. FASN is necessary for the expression of markers of glioma stem cells, and for the maintenance of stemness [40]. In this sense, the inhibition of FASN with the natural antibiotic cerulenin (Table 1) blocks cell proliferation and decreases the expression of the glioblastoma stemness markers Nestin, Sox2 and Fatty Acid Binding Protein 7 (FABP7) [40]. On the contrary, cerulenin increases the expression of the differentiation marker glial fibrillary acidic protein (GFAP). At last, FASN is overexpressed in induced pluripotent stem cells, neural stem and progenitor cells and glioma stem-like cells [41]: it is therefore likely that FASN is involved in the maintenance of stemness of CSCs (Fig. 3).
FASN and resistance to cell death
Migita et al. reported that the expression of FASN in human prostate tumor tissues was statistically significantly inverse to the apoptotic rate [13]. The activation of the intrinsic pathway of apoptosis with camptothecin in the prostate cancer LNCaP cells stably overexpressing FASN did not induce cell death contrary to control LNCaP cells [13] suggesting that FASN blocks the intrinsic pathway of apoptosis. In the same report, the authors showed that transgenic mice overexpressing FASN in the prostate harbored a prostatic hyperplasia associated with a weaker apoptotic rate compared to wild-type mice [13]. A study conducted on LNCaP cells demonstrated that knockdown of FASN favored apoptosis as evidenced by the increased-signal of Annexin V-EGFP and staining with propidium iodide. On the contrary, no effect was observed on fibroblasts viability [42].
Inhibition of FASN with C75 in HepG2 cells induces a G2-phase arrest associated with an increase of p53 expression [43]. Similarly, the treatment of PEL cells with C75 triggers apoptosis contrary to primary B cells [23]. The treatment of MDA-MB-231 cells with the natural FASN inhibitor curcumin (Table 1) or silencing FASN decreases the level of the anti-apoptotic protein Bcl-2 and concomitantly increases the pro-apoptotic protein Bax [44]. These observations strengthen the potent ability of FASN to oppose cancer cell apoptosis likely through their dependence upon DNL compared to healthy cells. Nevertheless, future investigations are necessary to decipher and understand the precise mechanism linking FASN to the extrinsic and intrinsic pathways of apoptosis at the molecular level.
FASN and cell invasion/metastasis
Jin et al. demonstrated that FASN was phosphorylated on Tyr66 by HER2 [36]. This phosphorylation activates FASN leading to cell invasion via an increase in the activity of the metalloproteinase MMP-9 (Fig. 4). Furthermore, FASN mediates the epithelial-mesenchymal transition (EMT) of breast cancer cells as its inhibition with cerulenin increases and decreases respectively the expression of E-cadherin and vimentin, and inhibits cell migration [45]. Hung et al. showed that the inhibition of FASN with the natural compound osthole (Table 1) or with C75 induced a loss of the oncogenic receptor c-Met inhibiting consequently EMT, migration and invasion of MCF-7 cells [46]. The deficit of c-Met was prevented by the addition of palmitate in the culture cell medium. Since c-Met is resident in lipid rafts [47] and that in epithelial cancer cells FASN activity is essential for structuring lipid rafts implicated in cell signaling and migration [48], this study suggests that the loss of FASN prevents c-Met localization in lipid microdomains (Fig. 4). It would be interesting to challenge the correct localization mechanism of c-Met at the level of lipid rafts in near future studies. Also, at the level of these membrane microdomains, FASN has been shown to interact with Src and Caveolin-1. Palmitoylation of caveolin-1, through the indirect activity of FASN, promotes the activation of Src and Akt which increases the migration of prostate cancer cells [49] (Fig. 4).

FASN is phosphorylated by HER2 increasing its activity and being responsible for an activation of MMP-9 that degrades the extracellular matrix (ECM). FASN exerts a positive feedback loop by allowing HER2 localization into lipid rafts. Through a respective reduction and increase of E-cadherin and vimentin expression, FASN promotes EMT reinforced by the proper localization of the c-Met receptor into lipid rafts. FASN interacts with Src and caveolin-1. FASN indirectly participates in caveolin-1 palmitoylation which in turn promotes Src and Akt activity leading to cell migration. The dotted arrows represent an indirect effect.
Expression of FASN in the high metastatic MHCC97H and SK-Hep-1 cell lines is increased compared with low metastatic HCC cell lines [19]. Transfection of HCC SK-Hep-1 cells with shFASN inhibited proliferation, migration and invasion [19]. Supplementation with palmitic acid improves HCC cell migration and activates the TGF-β and Wnt signaling pathways, both responsible for EMT program [50]. The DNL pathway promotes metastasis in mice; inhibiting FASN with the brain-permeable inhibitor BI99179 (Table 1) decreases the growth of brain metastases but does not impact the breast primary tumor [51]. In vivo, inhibiting the transcription factors sterol regulatory element binding proteins (SREBPs), that drive FASN expression, by fatostatin blocks both prostate tumor growth and distant metastasis [52]. Finally, cerulenin blocks cell migration and invasion of glioblastoma CSCs [40]. Consequently, FASN promotes metastasis, making the enzyme a prime target for preventing cancer progression and cancer cells spreading.
FASN and angiogenesis
FASN interferes with angiogenesis. The incubation of breast cancer cells with cerulenin results in a decrease expression of vascular endothelial growth factor (VEGF) and VEGFR-2 [45]. Moreover, FASN promotes the localization of VEGFR-2 on the endothelial cell surface [53]. It can be assumed that FASN promotes the localization of VEGFR-2 at the level of lipid rafts, and thus its activation by VEGF. but, to our knowledge this has never been clearly demonstrated (Fig. 5). In addition, Bruning et al. showed that knockdown of FASN in endothelial cells increased the malonylation of the kinase mTOR due to the excess of malonyl-CoA not used by FASN [54]. Malonylation inhibits mTOR resulting in a decrease in protein synthesis and to a loss of endothelial cell proliferation and angiogenesis (Fig. 5). Interestingly, the inhibition of FASN with orlistat (Table 1), a FDA-approved anti-obesity drug, or with cerulenin, significantly reduces metastases and angiogenesis in a mouse syngeneic model of melanoma [55]. In this model, FASN inhibition induced an increased secretion of an anti-angiogenic isoform of VEGF-A by melanoma cancer cells. Later, Zhou and collaborators showed that the expression level of FASN correlated with microvessels density in human gliomas [22]. They observed in mice orthotopically xenografted with GSCs that silencing FASN using shRNAs or inhibition with C75 decreased microvessels density and tumor growth. Mechanistically, the inhibition of FASN blocked hypoxia-inducible factor-1α (HIF-1α)/VEGF-A signaling and upregulated the anti-angiogenic isoform-VEGF-165b. But, the precise link between FASN and HIF-1α/VEGF-A signaling pathway deserves to be pushed a little further. Zaytseva et al. also demonstrated that the stable knockdown of FASN in colorectal cancer cells induced a decrease of angiogenesis [56]. The culture of human lung microvascular endothelial cells in a medium conditioned from FASN knockdown colorectal cancer cells decreased VEGFR-2 activation, cell proliferation, migration and tubulogenesis. These cancer cells secreted less MMP-9 and pro-angiogenic VEGF (VEGF-189) and conversely secreted more anti-angiogenic VEGF (VEGF-165b).

The catalytic activity of FASN probably allows the localization of the oncogenic receptor VEGFR-2 in lipid rafts, a prerequisite to the regulation of angiogenesis. By producing fatty acids, FASN reduces the pool of malonyl-CoA leading to a decrease of mTOR malonylation which promotes its activity and therefore angiogenesis. FASN also controls VEGF secretion and MMP-9 activation to respectively activate VEGFR-2 and degrade the extracellular matrix (ECM), and consequently to sustain angiogenesis. The expression and activity of the transcription factor HIF-1α are positively regulated by FASN; this induces VEGF-A expression. The dotted arrows represent an indirect effect.
FASN and immune escape
At last, several studies demonstrated a potential role of FASN in immune escape (Fig. 6). For instance, Sun et al. recently showed that the combined inhibition of FASN with C75 and of phosphatidylinositol 3-kinase-α (PI3Kα) with CYH33 synergistically enhances anti-tumoral immunity [57]. Following the injection of murine breast cancer cells 4T1 into the flank of immunocompetent BALB/c mice treated with C75 and CYH33, an increased apoptotic rate associated to a decrease in cancer cells proliferation was noticed. These observations were correlated to a respective increase and decrease of tumor infiltration by CD4+/CD8+ T lymphocytes and M2 macrophages. The combined inhibition of FASN and PI3Kα synergistically increased free fatty acids level in the tumor microenvironment. It is tempting to speculate that this increase could be partly responsible for the change observed in the composition in immune cells (Fig. 6).

Inhibition of FASN by orlistat induces NK cells and CD8+ T cells activation. Orlistat decreases the amount of T-regs whose suppressive activity is in part induced by FASN. Orlistat decreases PD-L1 expression, induces the secretion of nitric oxide (NO) by neutrophils and decreases the pool of fatty acids in dendritic cells which increases their maturation. The inhibition of FASN by C75 indirectly decreases the number of M2 macrophages but increases the pool of extracellular fatty acids that probably attract NK and CD4+/CD8+ T cells in the tumor microenvironnement. FASN is therefore involved in immune escape. The dotted arrows represent an indirect effect.
Another study focusing on regulatory T cells (T-regs) highlighted the involvement of FASN in immune suppression and therefore in immune escape [58]. Lim and collaborators showed that the activity of SREBPs was upregulated in intratumoral T-regs and promoted tumor growth and PD-1 expression. As FASN expression is promoted by SREBP-1, they engineered FASN-deficient T-regs cells and observed that these cells were less mature resulting in a decrease of tumor growth. The addition of palmitate in the medium of FASN-deficient T-regs cells restored the suppressive function of T-regs cells. These authors concluded that FASN signaling contributes to the functional maturation of T-regs cells indirectly via the activation of T cell antigen receptors (TCRs) [58] (Fig. 6).
Moreover, the treatment of a melanoma mouse model (B16-F10 cells/C57BL/6 mice) with orlistat reduced lymph node metastases reinforcing the involvement of FASN in immune escape [59]. Mechanistically, FASN inhibition increases the maturation of intratumoral dendritic cells, stimulates the expression of cytotoxicity markers of CD8+ T lymphocytes and natural killer cells and reduces the number of T-regs cells. In addition, blood neutrophils produce more nitric oxide which has antineoplastic effects (Fig. 6). Conversely, lipids produced by ovarian cancer cells overexpressing FASN accumulate in dendritic cells [60]. Dendritic cells are then unable to present the antigens to T lymphocytes and to induce an anti-tumor immune response. Finally, Cioccoloni et al. demonstrated that orlistat impairs the expression of PD-L1 implicated in immune suppression in a T-cell leukemia line [61]. This observation suggests that FASN promotes PD-L1 expression and, as a result, immune escape (Fig. 6).
General conclusion and perspectives
Due to its pivotal involvement in many features of cancer, FASN is potentially a good anti-cancer therapeutic target. To date, TVB-2640 (Table 1) is the most advanced FASN inhibitor currently used in phase 2 trial. Combined to taxane, TVB-2640 is promising in the treatment of advanced cancers [62]. This first-in-class inhibitor has been tested with patients carrying astrocytoma, NSCLC, breast cancer, peritoneal carcinoma and ovarian cancer [63]. TVB-3166 (Table 1), a smaller analogue of TVB-2640 used in vitro, leads to an endoplasmic reticulum stress which prevents the translation of ERα mRNAs in tamoxifen-resistant MCF-7 cells and results in a decrease of tumor growth of xenografted tumors in mice [64].
Of particular interest, PROteolysis TArgeting Chimeric molecules, or PROTACs, are promising tools that could be used to develop next-generation therapeutics to target FASN. PROTACs consist of chimeric molecules that bridge any protein to an E3 ligase so as to induce its proteasomal degradation. There are currently several PROTACs in clinical trials for the treatment of cancer such as ARV-471 (breast cancer) expected to enter phase 3 soon or ARV-110 (prostate cancer) which is in phase 2 [65]. These PROTACs are also combined to other drugs like Everolimus for ARV-471 (NCT05501769) or Abiraterone for ARV-110 (NCT05177042) in some clinical trials in an attempt to improve the treatment of patients. Thus, multi-compound therapies represent promising tools for the management of hard-to-cure cancer patients and could apply to FASN in conjunction with inhibitors currently in clinical phase.
FASN may also be responsible for the resistance to trastuzumab (Herceptin™) in HER2 overexpressing breast cancers. As discussed previously, there is a cross-talk between FASN and HER2 in which each promotes the expression of the other [36] (Figs. 3 and 4). The combination of trastuzumab with FASN blockade presents a synergistic effect on SK-BR-3 breast cancer cells growth inhibition and apoptosis [66]. Moreover, the inhibition of FASN-driven lipid rafts building also negatively affects EGFR-HER2 cross-talk which is of particular importance for trastuzumab resistance [48]. It has been also shown that tamoxifen promotes FASN expression in ER+/HER2+ breast cancer cells and increases cell proliferation [67]. Thus, FASN could also induce tamoxifen resistance by promoting cell growth. In NSCLC cells with mutated EGFR and resistant to TKI, the inhibition of FASN with orlistat induces EGFR K48-ubiquitination resulting in a reduction of tumor growth associated with an increase in apoptosis in mice xenografted tumors [39]. It would be particularly interesting to check whether this K48-ubiquitination of EGFR actually induces its proteasomal degradation.
FASN inhibition could be used as an adjuvant anti-cancer therapy to those already existing. For example, a synergistic effect of FASN and Thymidylate Synthase respectively inhibited by cerulenin and 5-fluorouracil is observed on cell viability in breast cancer cells [68]. In the same way, FASN blockade induces a synergistic chemo-sensitization of breast cancer cells to microtubule interfering agents such as docetaxel [69], paclitaxel [70] and vinorelbine [71]. As discussed previously the combined inhibition of FASN and PI3Kα synergistically inhibits tumor growth in a murine allograft model by enhancing anti-tumoral immunity [57]. As well, the inhibition of FASN in patients harboring a resistance to c-Met TKI is a promising therapy [46]. Finally, the FASN inhibitor G28 allows to overcome EGFR TKIs resistance in NSCLC [72]. The potential final approval of TVB-2640 or other anti-FASN drugs could therefore offer novel therapies for patients with cancer in an era of widespread resistance to existing drugs.
Data availability
All data presented in the current review are publicly available, and all findings summarized here come from articles cited in the reference list and available in the MEDLINE database.
References
Shurbaji MS, Kuhajda FP, Pasternack GR, Thurmond TS. Expression of oncogenic antigen 519 (OA-519) in prostate cancer is a potential prognostic indicator. Am J Clin Pathol. 1992;97:686–91. https://doi.org/10.1093/ajcp/97.5.686
Smith S, Witkowski A, Joshi AK. Structural and functional organization of the animal fatty acid synthase. Prog Lipid Res. 2003;42:289–317. https://doi.org/10.1016/s0163-7827(02)00067-x
Witkowski A, Ghosal A, Joshi AK, Witkowska HE, Asturias FJ, Smith S. Head-to-head coiled arrangement of the subunits of the animal fatty acid synthase. Chem Biol. 2004;1:1667–76. https://doi.org/10.1016/j.chembiol.2004.09.016
Raab S, Lefebvre T. L’acide gras synthase, une enzyme «multi-FASette». Med Sci. 2022;38:445–52. https://doi.org/10.1051/medsci/2022062
Maier T, Leibundgut M, Ban N. The crystal structure of a mammalian fatty acid synthase. Science. 2008;321:1315–22. https://doi.org/10.1126/science.1161269
Chirala SS, Chang H, Matzuk M, Abu-Elheiga L, Mao J, Mahon K, et al. Fatty acid synthesis is essential in embryonic development: fatty acid synthase null mutants and most of the heterozygotes die in utero. Proc Natl Acad Sci USA. 2003;100:6358–63. https://doi.org/10.1073/pnas.0931394100
Suburu J, Shi L, Wu J, Wang S, Samuel M, Thomas MJ. Fatty acid synthase is required for mammary gland development and milk production during lactation. Am J Physiol Endocrinol Metab. 2014;306:E1132–43. https://doi.org/10.1152/ajpendo.00514.2013
Wagle S, Bui A, Ballard PL, Shuman H, Gonzales, Gonzales LW. Hormonal regulation and cellular localization of fatty acid synthase in human fetal lung. Am J Physiol. 1999;277:L381–90. https://doi.org/10.1152/ajplung.1999.277.2.L381
Kuhla A, Blei T, Jaster R, Vollmar B. Aging is associated with a shift of fatty metabolism toward lipogenesis. J Gerontol A Biol Sci Med Sci. 2011;66:1192–200. https://doi.org/10.1093/gerona/glr124
Kuhajda FP, Jenner K, Wood FD, Hennigar RA, Jacobs LB, Dick JD, et al. Fatty acid synthesis: a potential selective target for antineoplastic therapy. Proc Natl Acad Sci USA. 1994;91:6379–83. https://doi.org/10.1073/pnas.91.14.6379
Vazquez-Martin A, Colomer R, Brunet J, Lupu R, Menendez JA. Overexpression of fatty acid synthase gene activates HER1/HER2 tyrosine kinase receptors in human breast epithelial cells. Cell Prolif. 2008;59–85. https://doi.org/10.1111/j.1365-2184.2007.00498.x
Rashid A, Pizer ES, Moga M, Milgraum LZ, Zahurak M, Pasternack GR, et al. Elevated expression of fatty acid synthase and fatty acid synthetic activity in colorectal neoplasia. Am J Pathol. 1997;150:201–8.
Migita T, Ruiz S, Fornari A, Fiorentino M, Priolo C, Zadra G, et al. Fatty acid synthase: a metabolic enzyme and candidate oncogene in prostate cancer. J Natl Cancer Inst. 2009;101:519–32. https://doi.org/10.1093/jnci/djp030
Kusakabe T, Nashimoto A, Honma K, Suzuki T. Fatty acid synthase is highly expressed in carcinoma, adenoma and in regenerative epithelium and intestinal metaplasia of the stomach. Histopathology. 2002;40:71–9. https://doi.org/10.1046/j.1365-2559.2002.01289.x
Orita H, Coulter J, Tully E, Abe M, Montgomery E, Alvarez H, et al. High levels of fatty acid synthase expression in esophageal cancers represent a potential target for therapy. Cancer Biol Ther. 2010;10:549–54. https://doi.org/10.4161/cbt.10.6.12727
Visca P, Sebastiani V, Botti C, Diodoro MG, Lasagni RP, Romagnoli F, et al. Fatty acid synthase (FAS) is a marker of increased risk of recurrence in lung carcinoma. Anticancer Res. 2004;24:4169–73.
Walter K, Hong SM, Nyhan S, Canto M, Fedarko N, Klein A, et al. Serum fatty acid synthase as a marker of pancreatic neoplasia. Cancer Epidemiol Biomark Prev. 2009;18:2380–5. https://doi.org/10.1158/1055-9965.EPI-09-0144
Cai Y, Wang J, Zhang L, Wu D, Yu D, Tian X, et al. Expressions of fatty acid synthase and HER2 are correlated with poor prognosis of ovarian cancer. Med Oncol. 2015;32:391. https://doi.org/10.1007/s12032-014-0391-z
Hao Q, Li T, Zhang X, Gao P, Qiao P, Li S, et al. Expression and roles of fatty acid synthase in hepatocellular carcinoma. Oncol Rep. 2014;32:2471–6. https://doi.org/10.3892/or.2014.3484
Raab S, Very N, Duchêne B, Rybarczyk P, Jonckheere N, El Yazidi-Belkoura I. et al. Evaluation of the expression of fatty acid synthase and O-GlcNAc transferase in patients with liver cancer by exploration of transcriptome databases and experimental approaches. Oncol Lett. 2022;23:105. https://doi.org/10.3892/ol.2022.13225.
Innocenzi D, Alò PL, Balzani A, Sebastiani V, Silipo V, La Torre G, et al. Fatty acid synthase expression in melanoma. J Cutan Pathol. 2003;30:23–8. https://doi.org/10.1034/j.1600-0560.2003.300104.x
Zhou Y, Jin G, Mi R, Zhang J, Zhang J, Xu H, et al. Inhibition of fatty acid synthase suppresses neovascularization via regulating the expression of VEGF-A in glioma. J Cancer Res Clin Oncol. 2016;142:2447–59. https://doi.org/10.1007/s00432-016-2249-6
Bhatt AP, Jacobs SR, Freemerman AJ, Makowski L, Rathmell JC, Dittmer DP, et al. Dysregulation of fatty acid synthesis and glycolysis in non-Hodgkin lymphoma. Proc Natl Acad Sci USA. 2012;109:11818–23. https://doi.org/10.1073/pnas.1205995109
Che L, Pilo MG, Cigliano A, Latte G, Simile MM, Ribback S, et al. Oncogene dependent requirement of fatty acid synthase in hepatocellular carcinoma. Cell Cycle. 2017;16:499–507. https://doi.org/10.1080/15384101.2017.1282586
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. https://doi.org/10.1016/j.cell.2011.02.013
Mounier C, Bouraoui L, Rassart E. Lipogenesis in cancer progression. Int J Oncol. 2014;45:485–92. https://doi.org/10.3892/ijo.2014.2441
Giudetti AM, De Domenico S, Ragusa A, Lunetti P, Gaballo A, Franck J, et al. A specific lipid metabolic profile is associated with the epithelial mesenchymal transition program. Biochim Biophys Acta Mol Cell Biol Lipids. 2019;1864:344–57. https://doi.org/10.1016/j.bbalip.2018.12.011
Geng F, Cheng X, Wu X, Yoo JY, Cheng C, Guo JY, et al. Inhibition of SOAT1 suppresses glioblastoma growth via blocking SREBP-1-mediated lipogenesis. Clin Cancer Res. 2016;22:5337–48. https://doi.org/10.1158/1078-0432.CCR-15-2973
Zhu Y, Gu L, Lin X, Zhou X, Lu B, Liu C, et al. USP19 exacerbates lipogenesis and colorectal carcinogenesis by stabilizing ME1. Cell Rep. 2021;37:110174. https://doi.org/10.1016/j.celrep.2021.110174
Southam AD, Khanim FL, Hayden RE, Constantinou JK, Koczula KM, Michell RH, et al. Drug redeployment to kill leukemia and lymphoma cells by disrupting scd1-mediated synthesis of monounsaturated fatty acids. Cancer Res. 2015;75:2530–40. https://doi.org/10.1158/0008-5472.CAN-15-0202
Calvisi DF, Wang C, Ho C, Ladu S, Lee SA, Mattu S, et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology. 2011;140:1071–83. https://doi.org/10.1053/j.gastro.2010.12.006
Guri Y, Colombi M, Dazert E, Hindupur SK, Roszik J, Moes S, et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell. 2017;32:807–23.e12. https://doi.org/10.1016/j.ccell.2017.11.011
Ravi D, Beheshti A, Abermil N, Lansigan F, Kinlaw W, Matthan NR, et al. Oncogenic integration of nucleotide metabolism via fatty acid synthase in non-hodgkin lymphoma. Front Oncol. 2021;11:725137. https://doi.org/10.3389/fonc.2021.725137
Raab S, Gadault A, Very N, Decourcelle A, Baldini S, Schulz C, et al. Dual regulation of fatty acid synthase (FASN) expression by O-GlcNAc transferase (OGT) and mTOR pathway in proliferating liver cancer cells. Cell Mol Life Sci. 2021;78:5397–413. https://doi.org/10.1007/s00018-021-03857-z
Lu T, Sun L, Wang Z, Zhang Y, He Z, Xu C. Fatty acid synthase enhances colorectal cancer cell proliferation and metastasis via regulating AMPK/mTOR pathway. OncoTargets Ther. 2019;1:3339–47. https://doi.org/10.2147/OTT.S199369
Jin Q, Yuan LX, Boulbes D, Baek JM, Wang YN, Gomez-Cabello D, et al. Fatty acid synthase phosphorylation: a novel therapeutic target in HER2-overexpressing breast cancer cells. Breast Cancer Res. 2010;12:R96. https://doi.org/10.1186/bcr2777
Rabionet M, Polonio-Alcalá E, Relat J, Yeste M, Sims-Mourtada J, Kloxin AM, et al. Fatty acid synthase as a feasible biomarker for triple negative breast cancer stem cell subpopulation cultured on electrospun scaffolds. Mater Today Bio. 2021;12:100155. https://doi.org/10.1016/j.mtbio.2021.100155
Yan C, Wei H, Minjuan Z, Yan X, Jingyue Y, Wenchao L, et al. The mTOR inhibitor rapamycin synergizes with a fatty acid synthase inhibitor to induce cytotoxicity in ER/HER2-positive breast cancer cells. PloS ONE. 2014;9:e97697. https://doi.org/10.1371/journal.pone.0097697
Ali A, Levantini E, Teo JT, Goggi J, Clohessy JG, Wu CS, et al. Fatty acid synthase mediates EGFR palmitoylation in EGFR mutated non-small cell lung cancer. EMBO Mol Med. 2018;10:e8313. https://doi.org/10.15252/emmm.201708313
Yasumoto Y, Miyazaki H, Vaidyan LK, Kagawa Y, Ebrahimi M, Yamamoto Y, et al. Inhibition of fatty acid synthase decreases expression of stemness markers in glioma stem cells. PLoS ONE. 2016;11:e0147717. https://doi.org/10.1371/journal.pone.0147717
Liu H, Zhang Z, Song L, Gao J, Liu Y. Lipid metabolism of cancer stem cells. Oncol Lett. 2022;23:1–8. https://doi.org/10.3892/ol.2022.13239
De Schrijver E, Brusselmans K, Heyns W, Verhoeven G, Swinnen JV. RNA interference-mediated silencing of the fatty acid synthase gene attenuates growth and induces morphological changes and apoptosis of LNCaP prostate cancer cells. Cancer Res 2003;63:3799–804.
Gao Y, Lin LP, Zhu CH, Chen Y, Hou YT, Ding J. Growth arrest induced by C75, A fatty acid synthase inhibitor, Was partially modulated by p38 MAPK but not by p53 in human hepatocellular carcinoma. Cancer Biol Ther. 2006;5:978–85. https://doi.org/10.4161/cbt.5.8.2883
Fan H, Liang Y, Jiang B, Li X, Xun H, Sun J, et al. Curcumin inhibits intracellular fatty acid synthase and induces apoptosis in human breast cancer MDA-MB-231 cells. Oncol Rep. 2016;35:2651–6. https://doi.org/10.3892/or.2016.4682
Li J, Dong L, Wei D, Wang X, Zhang S, Li H. Fatty acid synthase mediates the epithelial-mesenchymal transition of breast cancer cells. Int J Biol Sci. 2014;10:171–80. https://doi.org/10.7150/ijbs.7357
Hung CM, Kuo DH, Chou CH, Su YC, Ho CT, Way TD. Osthole suppresses hepatocyte growth factor (HGF)-induced epithelial-mesenchymal transition via repression of the c-Met/Akt/mTOR pathway in human breast cancer cells. J Agric Food Chem. 2011;59:9683–90. https://doi.org/10.1021/jf2021489
Duhon D, Bigelow RL, Coleman DT, Steffan JJ, Yu C, Langston W. The polyphenol epigallocatechin-3-gallate affects lipid rafts to block activation of the c-Met receptor in prostate cancer cells. Mol Carcinog. 2010;49:739–49. https://doi.org/10.1002/mc.20649
Menendez JA, Vellon L, Lupu R. Targeting fatty acid synthase-driven lipid rafts: a novel strategy to overcome trastuzumab resistance in breast cancer cells. Med Hypotheses. 2005;64:997–1001. https://doi.org/10.1016/j.mehy.2004.09.027
Di Vizio D, Adam RM, Kim J, Kim R, Sotgia F, Williams T, et al. Caveolin-1 interacts with a lipid raft-associated population of fatty acid synthase. Cell Cycle. 2008;7:2257–67. https://doi.org/10.4161/cc.7.14.6475
Nath A, Li I, Roberts LR, Chan C. Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Sci Rep. 2015;5:14752. https://doi.org/10.1038/srep14752
Ferraro GB, Ali A, Luengo A, Kodack DP, Deik A, Abbott KL, et al. Fatty acid synthesis is required for breast cancer brain metastasis. Nat Cancer. 2021;2:414–28. https://doi.org/10.1038/s43018-021-00183-y
Chen M, Zhang J, Sampieri K, Clohessy JG, Mendez L, Gonzalez-Billalabeitia E, et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat Genet. 2018;50:206–18. https://doi.org/10.1038/s41588-017-0027-2
Browne CD, Hindmarsh EJ, Smith JW. Inhibition of endothelial cell proliferation and angiogenesis by orlistat, a fatty acid synthase inhibitor. FASEB J. 2006;20:2027–35. https://doi.org/10.1096/fj.05-5404com
Bruning U, Morales-Rodriguez F, Kalucka J, Goveia J, Taverna F, Queiroz KCS, et al. Impairment of angiogenesis by fatty acid synthase inhibition involves mTOR malonylation. Cell Metab. 2018;28:866–80.e15. https://doi.org/10.1016/j.cmet.2018.07.019
Seguin F, Carvalho MA, Bastos DC, Agostini M, Zecchin KG, Alvarez-Flores MP, et al. The fatty acid synthase inhibitor orlistat reduces experimental metastases and angiogenesis in B16-F10 melanomas. Br J Cancer. 2012;107:977–87. https://doi.org/10.1038/bjc.2012.355
Zaytseva YY, Elliott VA, Rychahou P, Mustain WC, Kim JT, Valentino J, et al. Cancer cell-associated fatty acid synthase activates endothelial cells and promotes angiogenesis in colorectal cancer. Carcinogenesis. 2014;35:1341–51. https://doi.org/10.1093/carcin/bgu042
Sun P, Zhang X, Wang RJ, Ma QY, Xu L, Wang Y, et al. PI3Kα inhibitor CYH33 triggers antitumor immunity in murine breast cancer by activating CD8+T cells and promoting fatty acid metabolism. J Immunother Cancer. 2021;9:e003093. https://doi.org/10.1136/jitc-2021-003093
Lim SA, Wei J, Nguyen TM, Shi H, Su W, Palacios G, et al. Lipid signalling enforces functional specialization of Treg cells in tumours. Nature. 2021;591:306–11. https://doi.org/10.1038/s41586-021-03235-6
de Almeida LY, Mariano FS, Bastos DC, Cavassani KA, Raphelson J, Mariano VS, et al. The antimetastatic activity of orlistat is accompanied by an antitumoral immune response in mouse melanoma. Cancer Chemother Pharm. 2020;85:321–30. https://doi.org/10.1007/s00280-019-04010-1
Jiang L, Fang X, Wang H, Li D, Wang X. Ovarian cancer-intrinsic fatty acid synthase prevents anti-tumor immunity by disrupting tumor-infiltrating dendritic cells. Front Immunol. 2018;9:2927. https://doi.org/10.3389/fimmu.2018.02927
Cioccoloni G, Aquino A, Notarnicola M, Caruso MG, Bonmassar E, Zonfrillo M, et al. Fatty acid synthase inhibitor orlistat impairs cell growth and down-regulates PD-L1 expression of a human T-cell leukemia line. J Chemother. 2020;32:30–40. https://doi.org/10.1080/1120009X.2019.1694761
Lemberg KM, Gori SS, Tsukamoto T, Rais R, Slusher BS. Clinical development of metabolic inhibitors for oncology. J Clin Invest. 2022;132:e148550. https://doi.org/10.1172/JCI148550
Falchook G, Patel M, Infante J, Arkenau HT, Dean E, et al. Abstract CT153: first in human study of the first-in-class fatty acid synthase (FASN) inhibitor TVB-2640. Cancer Res. 2017;77:CT153. https://doi.org/10.1158/1538-7445.AM2017-CT153
Gruslova A, McClellan B, Balinda HU, Viswanadhapalli S, Alers V, Sareddy GR, et al. FASN inhibition as a potential treatment for endocrine-resistant breast cancer. Breast Cancer Res Treat. 2021;187:375–86. https://doi.org/10.1007/s10549-021-06231-6
Mullard A. Targeted protein degraders crowd into the clinic. Nat Rev Drug Disco. 2021;20:247–50. https://doi.org/10.1038/d41573-021-00052-4
Vazquez-Martin A, Colomer R, Brunet J, Menendez JA. Pharmacological blockade of fatty acid synthase (FASN) reverses acquired autoresistance to trastuzumab (Herceptin by transcriptionally inhibiting “HER2 super-expression” occurring in high-dose trastuzumab-conditioned SKBR3/Tzb100 breast cancer cells. Int J Oncol. 2007;31:769–76.
Menendez JA, Papadimitropoulou A, Vander Steen T, Cuyàs E, Oza-Gajera BP, Verdura S, et al. Fatty acid synthase confers tamoxifen resistance to ER+/HER2+ breast cancer. Cancers. 2021;13:1132. https://doi.org/10.3390/cancers13051132
Vazquez-Martin A, Ropero S, Brunet J, Colomer R, Menendez JA. Inhibition of Fatty Acid Synthase (FASN) synergistically enhances the efficacy of 5-fluorouracil in breast carcinoma cells. Oncol Rep. 2007;18:973–80.
Menendez JA, Lupu R, Colomer R. Inhibition of tumor-associated fatty acid synthase hyperactivity induces synergistic chemosensitization of HER -2/ neu -overexpressing human breast cancer cells to docetaxel (taxotere). Breast Cancer Res Treat. 2004;84:183–95. https://doi.org/10.1023/B:BREA.0000018409.59448.60
Menendez JA, Vellon L, Colomer R, Lupu R. Pharmacological and small interference RNA-mediated inhibition of breast cancer-associated fatty acid synthase (oncogenic antigen-519) synergistically enhances Taxol (paclitaxel)-induced cytotoxicity. Int J Cancer. 2005;115:19–35. https://doi.org/10.1002/ijc.20754
Menendez JA, Colomer R, Lupu R. Inhibition of tumor-associated fatty acid synthase activity enhances vinorelbine (Navelbine)-induced cytotoxicity and apoptotic cell death in human breast cancer cells. Oncol Rep. 2004;12:411–22.
Polonio-Alcalá E, Palomeras S, Torres-Oteros D, Relat J, Planas M, Feliu L, et al. Fatty acid synthase inhibitor G28 shows anticancer activity in EGFR tyrosine kinase inhibitor resistant lung adenocarcinoma models. Cancers. 2020;12:1283. https://doi.org/10.3390/cancers12051283
Author information
Authors and Affiliations
Contributions
DV drafted the manuscript, CS and TL edited the manuscript. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vanauberg, D., Schulz, C. & Lefebvre, T. Involvement of the pro-oncogenic enzyme fatty acid synthase in the hallmarks of cancer: a promising target in anti-cancer therapies. Oncogenesis 12, 16 (2023). https://doi.org/10.1038/s41389-023-00460-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41389-023-00460-8
This article is cited by
-
Lipid droplet accumulation in Wdr45-deficient cells caused by impairment of chaperone-mediated autophagic degradation of Fasn
Lipids in Health and Disease (2024)
-
The implications of FASN in immune cell biology and related diseases
Cell Death & Disease (2024)
