
Abstract
Post-traumatic stress disorder (PTSD) is a chronic incapacitating condition with recurrent experience of trauma-related memories, negative mood, altered cognition, and hypervigilance. Agglomeration of preclinical and clinical evidence in recent years specified that alterations in neural networks favor certain characteristics of PTSD. Besides the disruption of hypothalamus-pituitary-axis (HPA) axis, intensified immune status with elevated pro-inflammatory cytokines and arachidonic metabolites of COX-2 such as PGE2 creates a putative scenario in worsening the neurobehavioral facet of PTSD. This review aims to link the Diagnostic and Statistical Manual of mental disorders (DSM-V) symptomology to major neural mechanisms that are supposed to underpin the transition from acute stress reactions to the development of PTSD. Also, to demonstrate how these intertwined processes can be applied to probable early intervention strategies followed by a description of the evidence supporting the proposed mechanisms. Hence in this review, several neural network mechanisms were postulated concerning the HPA axis, COX-2, PGE2, NLRP3, and sirtuins to unravel possible complex neuroinflammatory mechanisms that are obscured in PTSD condition.
Similar content being viewed by others


Introduction
Post-traumatic stress disorder (PTSD) is a persistent enervative condition that progresses aftermath of a traumatic incident usually after the context of war, sexual violence, and natural disaster (American Psychological Association 2017). According to the 5th version of the Diagnostic and Statistical Manual of Mental Disorders (DSM-V), PTSD comprises four symptom clusters viz., presence of intrusive memories related to a stressful incident, persistent avoidance of stimuli, negative alteration in mood or cognition, and hyperarousal. These symptoms must have been present for one month after experiencing a traumatic event causing significant distress or social impairment. However, the symptoms may also be delayed even after years and the individual might experience a severe crisis later in life (Weathers et al. 2014; American Psychiatric Association 2017).
It is well documented that veterans and members of the armed forces are at greater risk of PTSD (Moore et al. 2021). The primary progenitor of PTSD among war veterans has certainly combated trauma (O’Toole et al. 2020). Around 8% of the general population suffers from posttraumatic stress disorder (PTSD). Veterans and active military personnel both experience PTSD at twice the rate as the general population (Judkins et al. 2020). According to WHO World Mental Health (WMH) surveys in 24 countries, rape (13.1%), sexual assault (15.1%), being stalked (9.8%), and sudden loss of a loved one (11.6%) had the largest proportions of this burden. The first three of these four traumas are very unusual with a high PTSD risk, whereas the fourth is a highly frequent event with a low PTSD risk (Kessler et al. 2017). Furthermore, the prevalence of PTSD in Indian settings varies greatly, from little to almost 70%. The disparity in PTSD prevalence rates has mostly been linked to methodological variations across the research, including variations in the severity of the catastrophe used for the study, technique of sampling, case identification, etc. (Pillai et al. 2016). Notably, a cross-sectional comparative study in Nepal identified strong association between pro-inflammatory cytokines and trauma, confirming the hypothesis of immune system activation in trauma (Koirala et al. 2023). Similarly, Case-control studies in PTSD patients showed high levels of chemokines and proinflammatory cytokines, indicating that these increased inflammatory markers could act as biomarkers of PTSD risk, resilience, and stress responses (Zhang et al. 2020; Otsuka et al. 2021). These upregulated proinflammatory markers imply that those suffering from PTSD may have an activated immune system, which might contribute to neuroinflammation (de Oliveira et al. 2018).
Neurobiological Mechanisms of Stress-Induced Inflammation in PTSD
Usually, the reaction to physiological and/or psychological stressors involves a coordinated interaction between autonomic and neuroendocrine responses (Morena et al. 2016). Stress stimulates the hypothalamic-pituitary-adrenal axis (HPA) to produce corticotrophin-releasing hormone (CRH) from paraventricular nucleus (PVN) of hypothalamus which fosters the anterior pituitary producing adrenocorticotropic hormone (ACTH) inducting glucocorticoid secretion (cortisol) to minimize immune responses (Hori and Kim 2019). Chronic stress is considered to jeopardize the progression of major depressive disorder (MDD), whereas exposure to acute yet extreme stressful events, often in individuals experiencing chronic stress, can precipitate the development of PTSD (Almeida et al. 2021).
A variety of studies have found hypocortisolism in people who have gone through a stressful event and then developed PTSD (Rohleder et al. 2004; Groer et al. 2015). Furthermore, expanding research has revealed that PTSD patients with hypocortisolism are more susceptible to developing proinflammatory cytokines (Daskalakis et al. 2016). Paraventriculare nucleus (PVN) neuronal activity in response to peripheral immunological challenge is mediated by cytokines-induced endothelial production of prostaglandins (Quan et al. 2003). This immune-induced HPA axis activation is primarily facilitated by prostaglandin E2 (PGE2) produced in the brain (Furuyashiki, Tomoyuki; Narumiya 2011). Notably, peripheral interleukin-1β (IL-1β) actuates the HPA axis, and increases ACTH release, likely through the initiation and release of CRH into the hypophyseal portal blood. PGE2 from endothelial cells of the brain microvasculature is also stimulated by peripheral IL-1β released from macrophages, which then acts PVN of the HPA axis (Parsadaniantz et al. 2000). Consequently, an increase in the release of CRH under chronic stress conditions and the further lack of a dearth of cortisol control over immune cells causes the, endothelial cells of PVN in the brain to mediate the immunological response and, exacerbating the PTSD condition.
The single prolonged stress (SPS) paradigm of PTSD in rats revealed that cyclooxygenase-2 (COX-2) can provoke inflammation and apoptosis in the hippocampus and contribute to the development of PTSD. Celecoxib, the selective COX-2 inhibitor decreased the levels of tumor necrosis factor- α (TNF-α), interleukin-6 (IL‐6), prostaglandin E2 (PGE2), and nitric oxide (NO) and hampered the neuronal apoptosis. Thus inhibition of COX-2 could decrease the occurrence of oxidative stress and apoptosis and can play a key role in clinical research and PTSD therapy in the future (Wang et al. 2018a). In LPS-challenged rats, doxycycline and meloxicam provided neuroprotection by lowering pro-inflammatory cytokine levels (TNF-α, IL-6, and IL-17) and COX-2 synthesis in the brain (Er et al. 2020). Moreover, studies using knockout mice and selective inhibitors have demonstrated that in a social defeat stress (SDS) model, toll receptors (TLR2/4), monoacylglycerol lipase (MAGL), and COX could induce PGE2 synthesis by TLR/MAGL/COX pathway causing social avoidance behavior (Nie et al. 2019).
As the global burden of Post-Traumatic Stress Disorder (PTSD) continues to rise and the disorder exhibits significant heterogeneity, it becomes crucial to comprehend the stress-related pathophysiology underlying PTSD. Though the existing research on the effectiveness of glucocorticoid-based treatment in preventing PTSD is encouraging, the mechanisms mediating this impact and the population that might benefit from this remains elusive due to methodological discrepancies and gaps in translational studies (Florido et al. 2023). Intervening in the established pathological pathways through repurposing drugs could offer a novel therapeutic approach to this complex condition (Table 1).
Channelization of Neural Systems Towards PTSD Symptomology
Serotonergic System, Inflammation, and PTSD
The physiological involvement of the neurotransmitter serotonin (5-hydroxytryptamine; 5-HT) is essential for brain development, mood regulation, and stress reactivity (Brummelte et al. 2017), whereas, a deficiency plays a pivotal role in the pathophysiology of depression (Jacobsen et al. 2012; Yohn et al. 2017), attention deficit hyperactivity disorder (ADHD) (Whitney et al. 2016) and Alzheimer’s disease (Chakraborty et al. 2019). Majority of the serotonin receptors are metabotropic, and produce their physiological responses through the second messenger systems and are G-protein-coupled receptors (GPCRs), whereas 5-HT3 is an ionotropic receptor and acts through ligand-gated ion channel (LGIC) (Sarkar et al. 2021).
Disruption of 5HT transmission has been proposed to be involved in the etiopathogenesis of PTSD. Low hippocampal 5HT levels have been linked with the development of depressive-like behavior in the SPS-induced stress model of PTSD in rats (Sherin and Nemeroff 2011; Lee et al. 2020). The concentration of 5HT in the dorsal median raphe is also reduced, which may alter the dynamics between the amygdala and the hippocampus (Sukhmanjeet Kaur Mann; Raman Marwaha. 2021). These results are in line with the PTSD condition, where low 5HT levels result in depressive behavior, impulsivity, and hypervigilance (Nisar et al. 2020). Furthermore, the time-dependent sensitization (TDS) stress in rats has shown that the considerably reduced plasma corticosterone levels lead to quantitative (receptor density) and qualitative (receptor affinity) alterations in the hippocampal 5HT1A and prefrontal cortex 5HT2A receptors (Harvey et al. 2003). Moreover, the density of 5HT1A receptors has also been shown to be upregulated in a rat model of PTSD suggesting the involvement of 5HT1A receptors in modulating the anxiety phenotype of PTSD (Luo et al. 2011), and these outcomes were consistent with the neuroimaging findings in PTSD patients (Sullivan et al. 2013). Overactivity of 5HT1B auto-receptors in dorsal raphe nucleus (DRN) neurons could be a key mediator of pathological reactions to stressful situations (Clark et al. 2002). In line with this, a genetic mouse model lacking 5-HT1B autoreceptors demonstrated lower anxiety-like behaviour in the open field and antidepressant-like effects in the forced swim and sucrose preference tests. These findings imply that strategies aiming at inhibiting 5-HT1B autoreceptors may be effective in the treatment of anxiety and depression which are the symptom clusters of PTSD (Nautiyal et al. 2016). The density of 5HT1B receptors has also been related to certain PTSD characteristics, implying that certain aspects of the clinical phenomenology of PTSD may be caused by these receptor changes (Bailey et al. 2013; Pietrzak et al. 2013b). A case-control genotyping study in adults who had experienced significant trauma found a link between genetic disparity in the 5HT2A promoter region and PTSD (Mellman et al. 2009). Further, chronic stress triggers the upregulation of 5HT2C receptors which have been linked to altered neuroplasticity and neuroinflammation due to the elevation of IL-6, IL-1β and calcinurin in PTSD (Règue et al. 2019). The reduction of serotonin transporter (SERT) gene expression is linked with contextual fear memory extinction in the SPS PTSD rat model, indicating that SERT attenuation is associated with stabilization of 5HT levels and inhibits hippocampus autophagy (Wu et al. 2016).
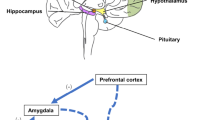
The HPA axis stress response is modulated by 5HT inputs from the DRN (Tafet and Nemeroff 2016) (Fig. 1), and direct synaptic interaction of serotonergic axons with CRH neurons and mediate the release of ACTH and cortisol through activation of 5HT1A, 5HT1B, 5HT2A, 5HT2B, and 5HT2C receptors in the hypothalamic PVN (Stephens and Wand 2012; Tung et al. 2012). The released cortisol has a detrimental impact on the DRN and 5HT-sensitive hippocampal neurons, which in turn results in low transcription of the gene encoding for 5HT1A receptor. 5HT1A auto receptor activation on the other hand results in DRN neuron hyperpolarization. This hyperpolarization decreases 5HT synthesis and altering the fear circuits and anxiety (Lanzenberger et al. 2010; Polter and Li 2010). Furthermore, corticosterone treatment in adrenalectomized rats has little effect on 5HT1B mRNA levels in the dorsal raphe or hippocampus as compared to 5HT1A mRNA (Neumaier et al. 2000). These results are significant since they hint at corticosterone’s selective modulation of receptors across brain areas. Evidence shows that genetic polymorphism in the central 5HT2A/2C receptors results in abnormal cortisol secretion after impaired motor control and attention (Brummett et al. 2012; Murnane 2019). Alongside receptor overexpression or polymorphism, low platelet serotonin levels and blunted HPA axis activity is also attributed to suicidality behavior in PTSD patients (Grah et al. 2010). These pieces of evidence reveal that abnormal HPA axis activation together with dysregulated serotonergic system contributes to negative symptoms of PTSD.

Neural networks modulating HPA axis: The stress response begins with the production of corticotropin-releasing hormone (CRH) from the HPA axis, which causes the anterior pituitary to produce adrenocorticotropic hormone (ACTH). Cortisol is then released by the action of ACTH on the adrenal cortex. It has negative feedback on the hypothalamus and anti-inflammatory effects. However, dysregulated neurotransmitters, along with hypocortisolism, may favour the development of neuroinflammation in PTSD (created by using Inkscape 1.2 version https://inkscape.org/)
Serotonin is involved in inflammatory and immunomodulatory disorders (Shajib and Khan 2015; Herr et al. 2017). Various in-vivo disease models and clinical investigations indicate that serotonergic transmission may influence the peripheral immune system. This raises serious concerns regarding the ability of various immune cells to produce, store, react to, and/or transport serotonin (Wu et al. 2019). The modulation of arachidonic acid (AA) turnover in the brain is instigated by 5HT via 5HT1A receptors (Strosznajder et al. 1994; Gopaldas et al. 2019) (Fig. 2). Various lines of evidence pointed out that 5HT2A receptor activation through Gα12/13 has been shown to control COX-2 activity as well as upsurge activation of serum-induced phospholipase A2 (PLA2) (Kurrasch-Orbaugh et al. 2003). Furthermore, selective 5HT2A/2 C receptor agonist 1-[2,5-dimethoxy-4-iodophenyl]-2-aminopropane (DOI), has been shown to induce COX-2 activation in the rat parietal cortex (Mackowiak et al. 2002) (Fig. 2). Although serotonin deficiency is one of the hallmarks in the pathophysiology of PTSD, explicit research is anticipated to confirm the contribution of the serotonergic system in the significant inflammatory imbalance scenario of PTSD. Taken together, altered 5HT transmission may cause PTSD symptoms such as hypervigilance, heightened startle, impulsivity, and intrusive memories (Fig. 3), however, the precise roles and mechanisms remain elusive.

Molecular mechanisms favoring neuroinflammation in PTSD: In the PTSD condition, dysregulated neurotransmitters interact differently at the receptor level, promoting the synthesis of cyclooxygenase-2 (COX-2), prostaglandin E2 (PGE2) and other proinflammatory cytokines. Furthermore, NOD-like receptor family pyrin domain-containing protein 3 (NLRP3) and Sirtuins (SIRT-1 and SIRT-6) may have a role in aggravating the development of neuroinflammation in the PTSD condition (created by using Inkscape 1.2 version https://inkscape.org/)

DSM-V clusters and their relationship with neurotransmitters: According to the Diagnostic and Statistical Manual of Mental Disorders (DSM-V), the four symptom clusters of PTSD include intrusive memories, avoidance of traumatic event reminders, negative mood and cognition, and hyperarousal. An imbalance between different neurotransmitters, such as excessive amounts of glutamate and noradrenaline and low levels of acetylcholine, γ-aminobutyric acid (GABA), dopamine, and serotonin encourage the development of these four PTSD symptom clusters (created by using Microsoft PowerPoint 2019)
Glutamatergic System, Inflammation, and PTSD
In the brain, glutamate is one of the major excitatory neurotransmitter and is at the crossroads of several metabolic pathways acts through both ionotropic (NMDA, AMPA, and kainate receptors), and/or metabotropic (mGluR1 − 8) glutamate receptors (Zhou and Danbolt 2014). Although most approaches indirectly evaluated glutamate neurosignaling, increasing evidence proposes glutamatergic dysfunction in several mental illnesses such as schizophrenia, Parkinson’s disease (PD), bipolar disorder (BD), major depressive disorder (MDD), obsessive-compulsive disorder (OCD), and PTSD (Li et al. 2019a; Nasir et al. 2020; Wang et al. 2020).
Glutamatergic signaling has a decisive role in the advancement of PTSD. Glutamate stimulates the release of CRH through moderation of neuronal inputs from the medial prefrontal cortex (mPFC) in the PVN of the hypothalamus, which modulates the HPA axis (Herman et al. 2002) (Fig. 1). Microinjections of glutamate into the rat PVN caused CRH and ACTH release, increased corticosterone levels, therefore favored arousal response by increasing the number of c-Fos positive CRH neurons (Kita et al. 2006). During the initial stage after trauma, elevated cortisol triggers the activation of NMDA receptors through NMDA-extracellular signal-regulated kinase (ERK) and mitogen and stress-activated kinase (MSK) (NMDA-ERK-MSK), and the glucocorticoid receptor (GR) pathways resulting in the formation of recurring memories, which is one of the phenotypes of PTSD (Fig. 3) (Reul and Nutt 2008). Neuroimaging studies and mGluR5 blockade studies have indicated that patients with PTSD overexpress mGluR5 and which is linked with suicidal ideation and avoidance symptoms (Holmes et al. 2017; Davis et al. 2019). In the SPS rat paradigm of PTSD, it was concluded that increased anxiety and impaired fear memory extinction were due to the overactivity of glutamatergic neurons with the subsequent difference between excitatory and inhibitory neurotransmission in the amygdala and this imbalance was correlated to the development of PTSD (Fang et al. 2018). These conclusions are in line with serum analysis and neuroimaging studies in PTSD patients, emphasizing the need for novel PTSD therapies that target the glutamatergic system. (Nishi et al. 2015; Harnett et al. 2017; Rosso et al. 2017).
A randomized controlled trial involving individuals who had surgery for minor head injuries revealed that selective COX-2 inhibition had a brain-protective effect via reducing glutamate levels (Bisri et al. 2017). In vitro studies also confirmed that glutamate NMDA receptor coupling results in Ca+ entry and propels neuronal nitric oxide synthase (nNOS) activation. The binding of nNOS to COX-2 results in NMDA-mediated excitotoxicity and also the formation of prostaglandins (Fig. 2). These outcomes suggest that the drugs inhibiting nNOS-COX-2 binding might lower prostaglandin levels in the brain, hence reducing excitotoxicity and neural dysfunctions. Secretory PLA2 (sPLA2) is stored in the synaptic vesicles and discharged in response to neural stimulation. In-vivo and in-vitro studies highlighted that both glutamate and sPLA2 increased the expression of neuronal COX-2, and these results indicate that COX-2 expression mediated by sPLA2 and glutamate results in the activation of the arachidonic acid paving the way for excitotoxicity-mediated neuroinflammation (Kolko et al. 1996, 2002).
GABAergic Neurotransmission and PTSD
GABA (gamma-aminobutyric acid) is a major inhibitory neurotransmitter in the CNS and is involved in regulating various physiological and pathophysiological pathways in the brain and peripheral tissues. GABA is largely produced from glutamine and glutamate by the action of glutaminase and glutamate decarboxylase (GAD) respectively (Watanabe et al. 2002). GABAergic neurons can be found in the hippocampus, thalamus, basal ganglia, hypothalamus, and brainstem. For optimal cell membrane integrity and neurologic function, a balance of inhibitory and excitatory neurotransmission via GABA and glutamate is required (Allen J, Mary; Sabir, Sarah; Sharma 2021).
GABAergic neurotransmission disruption may trigger PTSD pathogenesis (Arditte Hall et al. 2021) where the involvement of GABAA receptors is proven to inhibit hyperarousal state and anxiety. In juvenile rats, inescapable foot shock caused persistent anxiety, spatial memory loss, and decreased GABAAR subunit expression. Low brain levels of GABA are consistent with the overall findings in anxiety disorders and the hyperarousal hypothesis of both primary insomnia and PTSD (Meyerhoff et al. 2014). Low plasma concentration of GABA was seen in PTSD patients who also had symptoms of anxiety, avoidance, and hyperarousal suggesting that estimation of GABA levels might be used as a biomarker to assess PTSD severity (Fig. 3) (Trousselard et al. 2016). These results correspond to prior human neuroimaging research that indicates aberrant glutamate and GABA amounts in the brains of PTSD patients (Markus et al. 2016; Sheth et al. 2019).
While CRH neurons get inputs from a variety of brain regions, they are ultimately regulated by GABAergic inhibition from the amygdala (Myers et al. 2014) (Fig. 1) mediated by GABAAR on CRH neurons (Mody and Maguire 2012; Errington 2014). Electrophysiological recordings and in-vitro studies of CRH neurons in rodent hypothalamic brain slices have demonstrated that corticosteroids augment extrasynaptic GABAAR-mediated tonic currents (Herman et al. 2004; Colmers and Bains 2018). Moreover, selective deletion of the GABAA α1 subunit gene in the CRH neurons of mice produces a phenotype with increased anxiety as well as impaired fear memory extinction, both of which are hallmarks of PTSD (Gafford et al. 2012). Apart from differences in GABAAR expression, recent research indicates that the GABAergic neurotransmission in response to stress is more complicated, involving changes in chloride homeostasis as well as synaptic plasticity. Hence in the chronic stress disorders like PTSD, a condition of hypocortisolism, elevated CRH levels, downregulation of GABAAR with a substantial reduction in GABA levels favors hyperarousal (Fig. 3) and anxiety phenotypes of PTSD.
Activation of cyclooxygenases (COXs) may produce an increase in free radical generation, resulting in oxidative stress and apoptosis of GABAergic neurons and hence a rise in glutamate activity. Celecoxib, a selective COX-2 inhibitor has been shown to increase the expression of GABAA receptors and thereby increase the fast inhibitory neurotransmission in the hippocampus (Haiju et al. 2009). Whole-cell patch-clamp observations from parvocellular neuroendocrine cells (PNCs) of the hypothalamus in rats revealed that PGE2 inhibits the release of GABA onto para neuroendocrine cells (PNCs) in the PVN by presynaptic EP3 receptors, suggesting a possible mechanism by which local PGE2 activity in the PVN modulates the HPA axis during inflammation (Khazaeipool et al. 2018). Ferri and Ferguson in 2005 have shown that PNC cells in the PVN were depolarized by both IL-1β in a COX-2-dependent PGE2 stimulation, and the effect was dependent on the reduced GABAergic input caused by direct hyperpolarization of these neurons in the halo zone surrounding and projecting to the PVN (Ferri and Ferguson 2005) (Fig. 2). These evidence support the involvement of inhibitory amino acids involvement in PTSD and further detailed studies will add on to the existing understanding of this concept.
Dopaminergic System, Stress, Inflammation, and PTSD
Dopamine (DA) is a neurotransmitter that regulates motor control, motivation, reward, and cognitive function in CNS (Klein et al. 2019). In the mesocorticolimbic dopaminergic pathway, the synthesis of DA takes place in the midbrain ventral tegmental area (VTA) and is released into the nucleus accumbens (NAcc) and the medial prefrontal cortex (mPFC) (Juárez Olguín et al. 2016). All the dopaminergic receptors are metabotropic and D1, D2, D3, D4, and D5 receptors are linked to Gαs and Gαi G-proteins (Zhou et al. 2021).
Various studies have demonstrated that aberrant dopaminergic transmission from VTA to mPFC and hippocampus significantly contributes to PTSD symptoms including maladaptive memory consolidation (Torrisi et al. 2019; Zhou et al. 2021). Furthermore, the genetic alterations in the DA reuptake protein, dopamine transporter (DAT) could pave the way for PTSD condition (Drury et al. 2013; Zuschlag et al. 2021). Homozygous DAT gene ablation in rats showed neurodegeneration and glial cell activation providing new acumen into the association of DAT in the neuroinflammatory process (Illiano et al. 2021). Recently Yuan et al., (2021) have shown that Taq1A polymorphism of the DA D2 receptor (D2R), where the T allele carriers of D2R Taq1A express lower D2R density, higher levels of neuroinflammation and hippocampal atrophy and this causes reduced hippocampal subfield volume of CA3 region and severe PTSD symptoms (Yuan et al. 2021). In combat veterans with PTSD, the D2R gene is specifically linked to comorbid severe anxiety, depression, and social dysfunction (Lawford et al. 2006). Using a genome-wide DNA methylation pattern in a three-year follow-up study of veterans, it was shown that the Dopamine-PKA-CREB signaling pathway is often dysregulated and highly related to the hyperarousal phenotype in PTSD (Fig. 3) (Yang et al. 2020). Furthermore, deletion or antagonism of D3R resulted in anxiolytic effects in the rodent SPS model of PTSD, implying that D3 receptor antagonism was effective in reducing PTSD symptoms and also could decrease the risk of drug abuse and addiction (Rice et al. 2018; Song et al. 2018).
Although there are no direct dopaminergic projections from the VTA to the PVN, DA afferents from the VTA to the dorsolateral bed nucleus of stria terminalis (dlBNST) are crucial for the control of the HPA axis inhibiting CRH release (Di et al. 2020) (Fig. 1). Usually, under stress conditions, glucocorticoids stimulate DA release and generate euphoric emotions and movements in the mesolimbic dopaminergic regions (Butts and Phillips 2013; Howes et al. 2017). Various animal and clinical evidence emphasized that chronic stress impairs working memory via a significant reduction in DA concentration in the striatum and PFC (Mizoguchi et al. 2000; Bloomfield et al. 2019). Similarly, in the electric foot shock stress-restress exposure model of PTSD, it was shown that reduced hypothalamus and pituitary corticosterone levels, increased CRH, and glucocorticoid receptor gene expression (GR) were responsible for the reduced sniffing, rearing, and grooming activities in rats (Asalgoo et al. 2017). Furthermore, in SPS-induced stress in rats, low corticosterone is attributed to a decrease in DA levels along with elevated oxidative stress and neuroinflammation in cortical and hippocampal brain areas, which contributes to the clinical progression of PTSD (Uniyal et al. 2019). These findings imply that hypocortisolism and hypodopaminergia could potentially contribute to the negative mood and hyperarousal symptoms connected with PTSD (Fig. 3).
DA has been shown to modulate systemic inflammation through D1R signaling and acts as an endogenous inhibitor of the NLRP3 inflammasome activation pathway (Northrop and Yamamoto 2013). This suggests that DA may function as a coping mechanism against the development of inflammatory disorders and that D1R is a possible therapeutic target for NLRP3-driven inflammatory diseases (Yan et al. 2015; Wang et al. 2018b). Inflammatory cytokines have been shown to directly target DA and reward circuitry, contributing to depressive symptoms such as emotional exhaustion and motor retardation. Inflammatory cytokines appear to affect various elements of DA neurotransmission, resulting in reduced synthesis and/or defective packing or release, all of which may combine to varying degrees to lower DA function (Felger 2016). According to the current evidence, high DA levels activate low-affinity DA receptors (including D1R, D2R, and D4R), which have an anti-inflammatory impact on the cells of the immune system, but low DA levels precisely activate high-affinity DA receptors (including D3R and D5R), which causes neuroinflammation (Fig. 2). Under chronic stress conditions, as depicted in the chronic unpredictable stress paradigm in rats, COX activity caused an increase in striatal DAergic damage (Northrop and Yamamoto 2013). COX-2 is generally expressed at low levels in nigral DAergic neurons but it is up-regulated under both clinical and experimental conditions of Parkinson’s disease (Fathi Moghaddam et al. 2008). In animal models of Parkinson’s disease, COX-2 activation amplifies the cytotoxic effect by the activation of microglia and produces pro-inflammatory prostaglandins, iNOS, ROS and neurodegeneration (Chauhan et al. 2018; Ardah et al. 2020). Moreoveor, Carrasco et al., (2005) have shown that challenge of mesencephalic neuronal cultures by 6-OHDA and MPP + revealed, that exposure to 6-OHDA rather than MPP + in 24 h increased COX-2 dependent prostaglandin levels and Ibuprofen (COX inhibitor) suppressed PG rise and was inversely associated with dopaminergic cell death (Carrasco et al. 2005). COX-2 inhibition may protect from neuronal injury through microglia-independent mechanisms such as COX-2-mediated DA oxidation to quinone species, which produces oxidative stress and neuroinflammation (Chae et al. 2008; Vidal and Pacheco 2020). These data reinforce the use of anti-inflammatory drugs to treat neurodegenerative maladies involving DA and stress.
Adrenergic Neural Network and PTSD
The sympathetic nervous system controls a variety of biological activities including the regulation of the immune system (Sharma and Farrar 2020). Most of the noradrenergic neurons in CNS are localized in the brainstem nucleus, locus coeruleus (LC) (Berridge and Waterhouse 2003). LC neurons are involved in neuromodulation through neuronal inputs to the prefrontal cortex (PFC), basolateral amygdala (BLA), and motor cortex (Chandler et al. 2019). Noradrenaline (NA) release during acute stress generates a state of alertness and facilitates sensory processing to boost memory consolidation throughout stressful situations (Daviu et al. 2019).
Trauma and/or long-term stressors might produce dysregulation in noradrenergic neural networks, which has been implicated in the pathophysiology of PTSD as the source of hyperarousal clusters (Nwokafor et al. 2021). Neuroimaging findings in PTSD patients revealed that behavioral and autonomic hyper-responsiveness is induced by a strong phasic noradrenergic stimulus originating in the LC (Naegeli et al. 2018). Furthermore, cerebrospinal fluid (CSF) NA levels were shown to be high and positively linked with the severity of PTSD symptoms (Baker et al. 2001). There is an increased presynaptic outflow along with increased postsynaptic responsiveness to NA in CSF of PTSD patients (Geracioti et al. 2001). In-vivo studies and PET imaging in PTSD patients have indicated that considerable changes in norepinephrine transporter (NET) levels in the LC are related to an increase in the intensity of arousal symptoms (Pietrzak et al. 2013a; Sabban et al. 2018). In rodents, exaggerated acoustic startle response and reduced locomotor activity in a novel atmosphere have been utilized as indices of hyperarousal after a traumatic experience. Inescapable foot shock (IFS) enhanced the stress-induced extracellular concentration of NA in the amygdala and diminished locomotion as an indicator of hyperarousal in the PTSD condition (Jacek Dębiec, David E. A. Bush 2014; Ronzoni et al. 2016).
The release of CRH from CRH-containing terminals in the LC stimulates NA release (Jedema and Grace 2004) (Fig. 1). While increased noradrenergic activation of hypothalamic PVN may explain why CRH levels in PTSD patients are high. These data suggest that both acute and persistent upsurge in CRH outflow to the LC can boost noradrenergic outflow (O’Donnell et al. 2004). Further activation of CRH receptors may be altered by repeated stress-induced NA release (Rajbhandari and Bakshi 2020). Convergent models of PTSD show that cortisol and NA release leads to more intrusive memories in PTSD and the combination of NA and cortisol substantially predicts intrusive memories in PTSD patients. These findings imply a strong correlation between stress hormones and memory consolidation in PTSD requires a state of heightened arousal (Fig. 3) (Nicholson et al. 2014; Mather et al. 2016). Various clinical studies pointed out that patients with PTSD exhibited considerably increased NA secretion and decreased cortisol levels (Pervanidou 2012; Wingenfeld et al. 2015). This amplified NA release can provoke the synthesis of proinflammatory cytokines such as IL-1 and IL-6 via nuclear factor-κB (NF-κB)-dependent processes. Further excessive noradrenergic stimulation potentiates fear conditioning by inducing calcium influx in astrocytes via adrenergic receptors (Gazarini et al. 2013). Cortisol hinders sympathetic nervous system (SNS) hyperactivity via suppressing NF-κB signaling, which can reduce the production and release of proinflammatory cytokines (Tan et al. 2007). However, a persistent state of hypocortisolism in people with PTSD may lead to SNS hyperactivity, which accelerates inflammation.
In rats, ICV administration of arachidonic acid raised adrenaline and noradrenaline plasma levels after 20–30 min signifying that instigation of the brain phospholipase A2-arachidonic acid cascade promotes central sympatho-adrenomedullary outflow (Yokotani et al. 2000). Centrally administered CRH also enhances the expression of COX-1 and COX-2 in spinally projecting PVN neurons and COX-2 in LC neurons, indicating that COX isozymes are implicated in CRH-induced sympathetic modulation in rats (Yamaguchi and Okada 2009). Emerging evidence also suggests that PGE2 modulates sympathoexcitatory actions that are primarily arbitrated by the EP3 receptor (Zhang et al. 2011; Shimizu et al. 2014). These findings imply that the central excitatory effects of COX-2 and PGE2 on PVN neurons, in tandem with heightened sympathetic activity, hypocortisolism, and inflammation, may pave the way for neuroinflammation in PTSD.
Cholinergic System, Cognitive Inflexibility, and PTSD
Cholinergic signaling is critical for cognitive function, and its dysfunction is a hallmark of many neurodegenerative disorders, including Alzheimer’s disease (Hoskin et al. 2019; Winek et al. 2021). Furthermore, in various rodent stress models, cholinergic neurotransmission has been shown to play a crucial role in learning and memory extinction. (Srikumar et al. 2006; Yanpallewar et al. 2022). The hippocampus is enriched in cholinergic innervation and plays a pivtol role in cognitive function and stress-related behaviour (Pavlovsky et al. 2012). Acetylcholine (ACh) elicits its action through nicotinic and muscarinic receptors (Tiwari et al. 2013). Hence nicotine-based compounds have been proposed as potential therapeutical tools for the treatment of PTSD (Barreto et al. 2015). Cotinine, an active metabolite of nicotine has drawn significant attention in the recent past as a potential positive modulator of α7 nicotinic acetylcholine receptor (α7nAChR), and in a mice PTSD model, it actively enhances the fear extinction and reduces anxiety and depressive behaviour in an α7nAChR-dependent manner (Barreto et al. 2015; Mendoza et al. 2018; Aliev et al. 2020). Cotinine modulates synaptic plasticity and PTSD symptoms by stimulating downstream signaling of α7nAChR receptor; the protein kinase B (Akt)/glycogen synthase kinase 3β (GSK3β) pathway and ERKs (extracellular signal-regulated kinases) (Barreto et al. 2015; Mendoza et al. 2018). Furthermore, activation of α7nAChR modulates inflammatory pathways like, TLR4/NF-κB inflammasome and mTOR mediated autophagy and reduces pro-inflammatory cytokines (IL-6, IL-1β, TNF-α) (Bencherif et al. 2011; Ke et al. 2017). Consistent with these findings cotinine, a nootropic agent that modulates α7nAchR could be used as adjunctive therapy for PTSD (Table 1) and other neuropsychiatric conditions that cause neuroinflammation and dysfunction of learning and memory (Fig. 3) (Mendoza et al. 2018).
Reduced cognitive flexibility has recently been linked to predicting PTSD symptoms, where low flexibility has been suggested to be a risk factor for more severe PTSD symptoms (Ben-Zion et al. 2018). The cholinergic deficit has also been reported in the SRS-induced PTSD model in rodents, where donepezil ameliorated the SRS-induced cognitive inflexibility, downregulation of α7nAChR and expressed a reduced activity of choline acetyltransferase (ChAT) along with the increased activity of acetylcholine esterase (AChE) enzymes respectively (Prajapati and Krishnamurthy 2021). AChE activity in the basolateral amygdala in rats altered the strength or duration of cholinergic transmission during fear extinction (Kellis et al. 2020). In PTSD patients, single-photon emission computed tomography (SPECT) revealed that high concentrations of β2nAChRs in the thalamus promote re-experiencing symptoms through altering sensory input to the cortex and cortical neuroplasticity related to learning and stress response (Czermak et al. 2008). Various studies have also indicated that activation of α7nAChR lowers the levels of pro-inflammatory mediators and has a high potential to lower a variety of inflammatory-mediated ailments and neurological disorders, including PTSD (Bencherif et al. 2011; Sun et al. 2017; Ke et al. 2017). Inflammation, reduced baroreflex sensitivity (BRS), diminished parasympathetic nervous system (PNS), and excessive sympathetic nervous system (SNS) activity is proposed as contributory mechanisms for the severity of PTSD in a study including military veterans (Ulmer et al. 2018; Fonkoue et al. 2020).
Emerging data suggest that CRH modulates cognitive functions that rely on the cholinergic basal forebrain (Hupalo et al. 2019) through CRH1R present on cholinergic neurons and facilitates acetylcholine release (Day et al. 1998; Sauvage and Steckler 2001). Also, stress-induced responses activate the septohippocampal cholinergic pathway, which eventually activates the HPA axis (Paul et al. 2015) (Fig. 1). In bovine adrenal zona fasciculata/reticular (ZFR) cells, it was revealed that acetylcholine governs cortisol release at the cellular level via muscarinic M3 receptor (M3R) connected to phospholipase C (Walker et al. 1990). Stress in rodents enhanced the release of acetylcholine in the limbic areas (Imperato et al. 1989) and the HPA axis has been attributed to the susceptibility of basal forebrain cholinergic nerve cells (Aisa et al. 2009). Interestingly, Donepezil (AChE enzyme inhibitor) increased ACh availability, lowering negative symptoms in PTSD patients, and also inhibited LPS-induced neuroinflammation via α7nAChRs, which is followed by the PI3K-Akt mechanism, and this pathway might serve as a reference for the emergence of new therapies for reversing neuroinflammation or offer new indications for existing treatments (Table 1) (Tyagi et al. 2010; Navarro et al. 2021; Prajapati and Krishnamurthy 2021).
Cholinergic neurosignaling influences immune cell proliferation, cytokine production, T helper differentiation, and antigen presentation. These effects are facilitated through cholinergic muscarinic and nicotinic receptors and other cholinergic constituents found in immune cells, such as AChE and ChAT. Acetylcholine protects neurons from LPS-induced neuronal damage by suppressing the inflammatory response in rats (Li et al. 2019b). The anti-inflammatory mechanism favored by α7nAChR activation occurs through recruitment and stimulation of the Jak2/STAT3 pathway, which suppresses NF-κB nuclear translocation (Fig. 2) while activating the master regulator of oxidative stress Nrf2/HO-1 (Egea et al. 2015; Patel et al. 2017). Recent rodent experiments revealed that activation of α7nAChR reduced COX-2 expression, microsomal prostaglandin E synthase-1 (mPGES-1), and secretion of PGE2 (Piovesana et al. 2021; Peng-Fei et al. 2021). Nevertheless, in PTSD, hypoactivity of the cholinergic system with downregulated α7nAChR receptors could favor the negative mood and cognition aggravated with an inflammatory condition (Fig. 2).
NLRP3 Inflammasomes and PTSD
Emerging evidence of research emphasized the menacing role of NOD-like receptor family pyrin domain-containing protein 3 (NLRP3) in the etiology of many neurodegenerative disorders (Holbrook et al. 2021), traumatic brain and spinal cord injury (Zhou et al. 2022) and neuroinflammation (Lin and Mei 2021). The NLRP3 inflammasome is a multimeric protein complex that initiates pyroptosis and causes proinflammatory cytokines to be released (Yang et al. 2019b). It is made up of a sensor (NLRP3), an adapter (ASC; also known as PYCARD) and an effector (caspase 1). NLRP3 contains amino-terminal pyrin domain (PYD), a core NACHT domain (Swanson et al. 2019), combined with the adapter molecule apoptosis-associated speck-like protein comprising CARD (ASC) to recruit the effector caspase-1 to allow the IL-1 family cytokines IL-1β and IL-18 to be proteolytically cleaved. TLR agonists induce NF-κB-mediated NLRP3 formation and pro-IL-1β expression (priming phase) along with ATP, K+ ionophores, heme, and pathogen-associated RNA promotes NLRP3 inflammatory assembly (activation) caspase-1-mediated IL-1β, IL-18 secretion, and pyroptosis (Yang et al. 2019b). In addition to these upstream activities, a variety of NLRP3-interacting proteins and posttranslational changes to NLRP3 often control inflammatory activation of NLRP3 (Duan et al. 2020).
There has been limited literature supporting the direct involvement of NLRP3 in PTSD. However, it has recently been shown in the SPS-induced PTSD rat model that inhibition of NLRP3 inflammasome activity by using an endogenous inhibitor, β-hydroxybutyrate (BHB) produces anxiolytic effects and reduced stress-induced TNF-α levels (Yamanashi et al. 2020). These findings imply that administering BHB might effectively tackle the inflammatory pathways linked with PTSD (Yamanashi et al. 2020). Similarly, deletion of the NLRP3 gene in mice demonstrated that the NLRP3 inflammasome was stimulated in the hippocampus 72 h following electric foot shocks in a contextual fear paradigm, which was accompanied by an increase in the toll-like receptor, retinoic acid-inducible gene (RIG-I) like receptor signaling, and a decrease in post synaptic density (PSD)-related proteins. Both genetic deletion and pharmacologic blockade of the NLRP3 inflammasome may improve extinction of contextual fear memory and reduce anxiety-like behavior, offering a novel therapy for trauma and stress-related disorders such as PTSD (Dong et al. 2020). In-silico and in-vitro studies employing phenylpropanoids performed in our lab, have yielded encouraging results against neuroinflammation. The compounds studied strongly suppressed the NLRP3 inflammasome pathway in glial cells, as shown by mRNA levels of key proteins and IL-1β production (Kinra et al. 2021).
In-vitro and in-vivo studies conducted by (Feng et al. 2019) revealed that chronic stress activates the GR-NF-κB-NLRP3 signaling in microglia, causing hippocampal neuroinflammation and depression-like behavior. Chronic stress also causes glucocorticoid resistance as observed in PTSD permits proinflammatory signaling pathways markedly by IL-1β which is a byproduct of NLRP3 activation, to bypass normal feedback control. The surge in IL-1β, in particular, may not be counteracted by a deficiency of cortisol, and this incidence might destabilize the CNS (Zefferino et al. 2021). NLRP3 is also engaged in serotonergic (Iwata et al. 2013), Glutamatergic (Yang et al. 2019a), GABAergic (Zhang et al. 2013; Xia et al. 2021), Dopaminergic (Yan et al. 2015), Adrenergic (Horstmann et al. 2016) and cholinergic (Ke et al. 2017; Wei et al. 2019) systems of CNS.
IL-1β can stimulate gene expression and production of COX-2 and PGE2 (Dinarello 2009). It has been proposed that COX-2 has a crucial role in PGE2-induced activation of the NLRP3 inflammasome, which is mediated via activation of NF-κB (Fig. 2) and caspase-1, as well as the release of mtDNA and mtROS. Furthermore, in response to the LPS-challenge, COX-2 inhibition in mice with celecoxib lowered IL-1β and caspase-1 in the spleen and liver. These findings provide novel insights on how COX-2 controls NLRP3 inflammasome activation and indicate that it might be a novel potential therapeutic target (Table 1) in NLRP3-related illnesses (Zhang et al. 2018; Hung et al. 2019).
Sirtuins and PTSD
Sirtuins (SIRTs) are ubiquitous regulators of cell activities that are class III histone deacetylases and have been shown to have neuroprotective properties in a variety of neurodegenerative disorders (Liu et al. 2021; Ranadive et al. 2021). In PVN, SIRT1 stimulates the HPA axis and basal glucocorticoid (GC) levels by increasing CRH production via an increase in prohormone convertase 2 (PC2) biosynthesis, which is required for the maturation of CRH from pro-CRH (Toorie et al. 2016; Yamamoto and Takahashi 2018). SIRT1 induces deacetylation of helix loop helix transcription factor 2 (NHLH2) in the ventral CA1 region of the brain and enhances MAO-A transcription which then results in decomposition of serotonin (5HT) to 5 hydroxy indole acetic acid (5-HIAA) and influences PTSD-like moods and behaviors in SPS model of PTSD. While SIRT1 knockout mice and its inhibitor exhibited diminished anxiety and fear memory behaviors following the SPS procedure. These findings suggested that SIRT1 may be associated with the development of PTSD-like symptoms in reaction to extreme stress (Libert et al. 2011). Enhanced contextual memory is persistently noticed in PTSD (Al Abed et al. 2020). In the brain, ventral CA1 (vCA1) hippocampal projections deliver aversive stimuli-related inputs to the basal amygdala (BA), which serves to encode conditioned fear memory (Kim and Cho 2020). Furthermore, loss of SIRT6 in neuronal progenitors leads to tau-protein accumulation and loss of associative and non-associative memory, whereas overexpression of SIRT6 impairs long-term contextual fear memory by hindering IGF/Akt signalling pathway, that stimulates cAMP response element-binding protein (CREB). This pathway may be activated and contributed to the increase of contextual fear memory (Yin et al. 2016; Kaluski et al. 2017). Interestingly, genetic SIRT6 depletion in excitatory neurons, showed significant elevated contextual fear memory while spatial memory was not effected suggesting that enhancement in negative memory was due to reduced SIRT6 activity (Kim et al. 2018).
In the human umbilical vein endothelial cells (HUVECs), SIRT6 overexpression was associated with reduced NF-κB transcriptional activity, whereas knockdown of SIRT6 boosted NF-κB expression and resulted in COX-2, PGE2, and pro-inflammatory cytokines production (IL-6, IL-8) (Fig. 2). The overall outcomes of this study reveals that the loss of SIRT6 in endothelial cells is connected with an increase in the expression of genes implicated in inflammation (Lappas 2012). Although sirtuins are involved in alleviating neurodegenerative disorders (Yeong et al. 2020), SIRT 1 paradoxically increases the risk of PTSD while SIRT6 indirectly reduces PTSD symptoms. These findings further warrant more research to establish the detrimental or protective role of sirtuins in PTSD.
Conclusion
The pharmacological treatment of PTSD has been restricted due to the narrow focus on the monoamine system and a lack of efficacy with current treatment approaches, indicating a gap in translating basic research to clinical research. Thus, future research on the pharmacological treatment of PTSD should not only concentrate on novel neurotransmitter pathways but also aim to enhance the understanding of the pathophysiology of the cognitive and emotional processes involved in PTSD. This will help to fully restore functions rather than merely compensating for posited deficits, leading to therapeutic innovations in the field of PTSD. This review summarises the psycho-neuro-immunological interplay in PTSD which could pave the way for neuroinflammation. Hence parallel to mechanistic research, efforts should be aimed at identifying novel pathways that would unravel the new treatment outcome is an emerging challenge that could lead to effective methods of preventing and treating PTSD.
Availability of Data and Material (data transparency)
Not applicable.
Code Availability (software application or custom code)
Not applicable.
References
Aisa B, Gil-Bea FJ, Marcos B et al (2009) Neonatal stress affects vulnerability of cholinergic neurons and cognition in the rat: involvement of the HPA axis. Psychoneuroendocrinology 34:1495–1505. https://doi.org/10.1016/j.psyneuen.2009.05.003
Al Abed AS, Ducourneau EG, Bouarab C et al (2020) Preventing and treating PTSD-like memory by trauma contextualization. Nat Commun 11:1–9. https://doi.org/10.1038/s41467-020-18002-w
Aliev G, Beeraka NM, Nikolenko VN et al (2020) Neurophysiology and psychopathology underlying PTSD and recent insights into the PTSD therapies—a comprehensive review. J Clin Med 9:1–19
Allen J, Sabir M, Sharma S (2021) S GABA Receptor. In:tatPearls. https://www.ncbi.nlm.nih.gov/books/NBK526124/. Accessed 6 Aug 2021
Almeida FB, Barros HMT, Pinna G (2021) Neurosteroids and neurotrophic factors: what is their promise as biomarkers for major depression and PTSD? Int J Mol Sci 22:1–12. https://doi.org/10.3390/ijms22041758
American Psychiatric Association (2017) DSM-5 update. Diagnostic and statistical manual of mental disorder
American Psychological Association (2017) Clinical practice Guideline for the treatment of posttraumatic stress disorder (PTSD). APA, Washington, DC. https://www.apa.org/ptsd-guideline/ptsd.pdfhttps://www.apa.org/about/offices/directorates/guidelines/ptsd.pdf Guideline Development Panel for the Treatment of Posttraumatic Stress Disorder in Adults
Ardah MT, Bharathan G, Kitada T, Haque ME (2020) Ellagic acid prevents dopamine neuron degeneration from oxidative stress and neuroinflammation in MPTP model of Parkinson’s disease. Biomolecules 10:1–17. https://doi.org/10.3390/biom10111519
Arditte Hall KA, DeLane SE, Anderson GM et al (2021) Plasma gamma-aminobutyric acid (GABA) levels and posttraumatic stress disorder symptoms in trauma-exposed women: a preliminary report. Psychopharmacology 238:1541–1552. https://doi.org/10.1007/s00213-021-05785-z
Asalgoo S, Tat M, Sahraei H, Jahromi GP (2017) The psychoactive agent crocin can regulate hypothalamic-pituitary-adrenal axis activity. Front Neurosci 11:1–10. https://doi.org/10.3389/fnins.2017.00668
Astill Wright L, Sijbrandij M, Sinnerton R et al (2019) Pharmacological prevention and early treatment of post-traumatic stress disorder and acute stress disorder: a systematic review and meta-analysis. Transl Psychiatry 9:1–10. https://doi.org/10.1038/s41398-019-0673-5
Aykac A, Şehirli A, Gören MZ (2020) Evaluation of the Effect of Prazosin Treatment on α-2c adrenoceptor and apoptosis protein levels in the Predator Scent-Induced rat model of post-traumatic stress disorder. J Mol Neurosci. https://doi.org/10.1007/s12031-020-01518-7
Bailey CR, Cordell E, Sobin SM, Neumeister A (2013) Recent progress in understanding the pathophysiology of post-traumatic stress disorder. CNS Drugs 27:221–232. https://doi.org/10.1007/s40263-013-0051-4
Baker DG, Ekhator NN, West SA et al (2001) CSF norepinephrine concentrations in posttraumatic stress disorder.American Journal of Psychiatry1227–1230
Baptista-de-Souza D, Tavares LRR, Furuya-da-Cunha EM et al (2020) Chronic Fluoxetine impairs the Effects of 5-HT1A and 5-HT2C receptors activation in the PAG and Amygdala on Antinociception Induced by Aversive Situation in mice. Front Pharmacol 11:1–14. https://doi.org/10.3389/fphar.2020.00260
Barreto G, Yarkov A, Avila-Rodriguez M et al (2015) Nicotine-derived Compounds as therapeutic tools against post-traumatic stress disorder. Curr Pharm Des 21:3589–3595. https://doi.org/10.2174/1381612821666150710145250
Ben-Zion Z, Fine NB, Keynan NJ et al (2018) Cognitive flexibility predicts PTSD symptoms: observational and interventional studies. Front Psychiatry 9. https://doi.org/10.3389/fpsyt.2018.00477
Bencherif M, Lippiello PM, Lucas R, Marrero MB (2011) Alpha7 nicotinic receptors as novel therapeutic targets for inflammation-based diseases. Cell Mol Life Sci 68:931–949. https://doi.org/10.1007/s00018-010-0525-1
Berridge CW, Waterhouse BD (2003) The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Rev 42:33–84. https://doi.org/10.1016/S0165-0173(03)00143-7
Bisri DY, Arifin Z, Redjeki IS et al (2017) The effect of selective COX-2 inhibitor on blood glutamate in moderate traumatic brain injury. Crit Care Shock 20:30–39
Bloomfield MA, McCutcheon RA, Kempton M et al (2019) The effects of psychosocial stress on dopaminergic function and the acute stress response. Elife 8:1–22. https://doi.org/10.7554/eLife.46797
Brummelte S, Mc Glanaghy E, Bonnin A, Oberlander TF (2017) Developmental changes in serotonin signaling: implications for early brain function, behavior and adaptation. Neuroscience 342:212–231. https://doi.org/10.1016/j.neuroscience.2016.02.037
Brummett BH, Kuhn CM, Boyle SH et al (2012) Cortisol responses to emotional stress in men: Association with a functional polymorphism in the 5HTR2C gene. Biol Psychol 89:94–98. https://doi.org/10.1016/j.biopsycho.2011.09.013
Butts KA, Phillips AG (2013) Glucocorticoid receptors in the prefrontal cortex regulate dopamine efflux to stress via descending glutamatergic feedback to the ventral tegmental area. Int J Neuropsychopharmacol 16:1799–1807. https://doi.org/10.1017/S1461145713000187
Carrasco E, Casper D, Werner P (2005) Dopaminergic neurotoxicity by 6-OHDA and MPP+: Differential requirement for neuronal cyclooxygenase activity. J Neurosci Res 81:121–131. https://doi.org/10.1002/jnr.20541
Chae SW, Kang BY, Hwang O, Choi HJ (2008) Cyclooxygenase-2 is involved in oxidative damage and alpha-synuclein accumulation in dopaminergic cells. Neurosci Lett 436:205–209. https://doi.org/10.1016/j.neulet.2008.03.031
Chakraborty S, Lennon JC, Malkaram SA et al (2019) Serotonergic system, cognition, and BPSD in Alzheimer’s disease. Neurosci Lett 704:36–44
Chandler DJ, Jensen P, McCall JG et al (2019) Redefining Noradrenergic Neuromodulation of Behavior: impacts of a modular locus Coeruleus Architecture. J Neurosci 39:8239–8249. https://doi.org/10.1523/JNEUROSCI.1164-19.2019
Chauhan AK, Mittra N, Patel DK, Singh C (2018) Cyclooxygenase-2 directs microglial activation-mediated inflammation and oxidative stress leading to intrinsic apoptosis in Zn-Induced parkinsonism. Mol Neurobiol 55:2162–2173. https://doi.org/10.1007/s12035-017-0455-0
Cisler JM, Privratsky AA, Sartin-Tarm A et al (2020) l-DOPA and consolidation of fear extinction learning among women with posttraumatic stress disorder. Transl Psychiatry 10. https://doi.org/10.1038/s41398-020-00975-3
Clark MS, Sexton TJ, McClain M et al (2002) Overexpression of 5-HT1B receptor in dorsal Raphe Nucleus using herpes simplex virus gene transfer increases anxiety behavior after inescapable stress. J Neurosci 22:4550–4562. https://doi.org/10.1523/jneurosci.22-11-04550.2002
Colmers PLW, Bains JS (2018) Balancing tonic and phasic inhibition in hypothalamic corticotropin-releasing hormone neurons. J Physiol 596:1919–1929. https://doi.org/10.1113/JP275588
Corchs F, Nutt DJ, Hood S, Bernik M (2009) Serotonin and sensitivity to trauma-related exposure in selective serotonin reuptake inhibitors-recovered posttraumatic stress disorder. Biol Psychiatry 66:17–24. https://doi.org/10.1016/j.biopsych.2009.01.031
Czermak C, Staley JK, Kasserman S et al (2008) β2 nicotinic acetylcholine receptor availability in post-traumatic stress disorder. Int J Neuropsychopharmacol 11:419–424. https://doi.org/10.1017/S1461145707008152
Daskalakis NP, Cohen H, Nievergelt CM et al (2016) New translational perspectives for blood-based biomarkers of PTSD: from glucocorticoid to immune mediators of stress susceptibility. Exp Neurol 284:133–140. https://doi.org/10.1016/j.expneurol.2016.07.024
Davis MT, Hillmer A, Holmes SE et al (2019) In vivo evidence for dysregulation of mGluR5 as a biomarker of suicidal ideation. Proc Natl Acad Sci 116:11490–11495. https://doi.org/10.1073/pnas.1818871116
Daviu N, Bruchas MR, Moghaddam B et al (2019) Neurobiological links between stress and anxiety. Neurobiol Stress 11:100191. https://doi.org/10.1016/j.ynstr.2019.100191
Day JC, Koehl M, Deroche V et al (1998) Prenatal stress enhances stress- and corticotropin-releasing factor- induced stimulation of hippocampal acetylcholine release in adult rats. J Neurosci 18:1886–1892. https://doi.org/10.1523/jneurosci.18-05-01886.1998
de Moraes Costa G, Zanatta FB, Ziegelmann PK et al (2020) Pharmacological treatments for adults with post-traumatic stress disorder: a network meta-analysis of comparative efficacy and acceptability. J Psychiatr Res 130:412–420
de Oliveira JF, Wiener CD, Jansen K et al (2018) Serum levels of interleukins IL-6 and IL-10 in individuals with posttraumatic stress disorder in a population-based sample. Psychiatry Res 260:111–115. https://doi.org/10.1016/j.psychres.2017.11.061
Di T, Wang Y, Zhang Y et al (2020) Dopaminergic afferents from midbrain to dorsolateral bed nucleus of stria terminalis inhibit release and expression of corticotropin-releasing hormone in paraventricular nucleus. J Neurochem 154:218–234. https://doi.org/10.1111/jnc.14992
Dinarello CA (2009) Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 27:519–550. https://doi.org/10.1146/annurev.immunol.021908.132612
Dong Y, Li S, Lu Y et al (2020) Stress-induced NLRP3 inflammasome activation negatively regulates fear memory in mice. J Neuroinflammation 17:1–16. https://doi.org/10.1186/s12974-020-01842-0
Drury SS, Brett ZH, Henry C, Scheeringa M (2013) The association of a novel haplotype in the dopamine transporter with preschool age posttraumatic stress disorder. J Child Adolesc Psychopharmacol 23:236–243. https://doi.org/10.1089/cap.2012.0072
Duan Y, Kelley N, He Y (2020) Role of the NLRP3 inflammasome in neurodegenerative diseases and therapeutic implications. Neural Regen Res 15:1249–1250. https://doi.org/10.4103/1673-5374.272576
Egea J, Buendia I, Parada E et al (2015) Anti-inflammatory role of microglial alpha7 nAChRs and its role in neuroprotection. Biochem Pharmacol 97:463–472. https://doi.org/10.1016/j.bcp.2015.07.032
Er A, Coskun D, Bahcivan E, Dik B (2020) Effect of doxycycline and meloxicam on cytokines, brain-derived neurotrophic factor, matrix metalloproteinase-3, tissue inhibitor of metalloproteinase-3 and cyclooxygenase-2 in brain. Iran J Basic Med Sci 23:1328–1334. https://doi.org/10.22038/ijbms.2020.45193.10527
Errington CA (2014) Extrasynaptic GABAA receptors. Springer New York, New York, NY
Fang Q, Li Z, Huang G, di et al (2018) Traumatic stress produces distinct activations of GABAergic and glutamatergic neurons in amygdala. Front Neurosci 12:1–13. https://doi.org/10.3389/fnins.2018.00387
Fathi Moghaddam H, Ardestani MS, Saffari M et al (2008) Dopaminergic but not glutamatergic neurotransmission is increased in the striatum after selective cyclooxygenase-2 inhibition in normal and hemiparkinsonian rats. Basic Clin Pharmacol Toxicol 103:293–296. https://doi.org/10.1111/j.1742-7843.2008.00295.x
Felger JC (2016) The Role of Dopamine in Inflammation-Associated Depression: Mechanisms and Therapeutic Implications. In: Brain Imaging in Behavioral Neuroscience. pp 199–219
Feng X, Zhao Y, Yang T et al (2019) Glucocorticoid-driven NLRP3 inflammasome activation in hippocampal Microglia mediates chronic Stress-Induced Depressive-Like Behaviors. Front Mol Neurosci 12. https://doi.org/10.3389/fnmol.2019.00210
Ferri CC, Ferguson A (2005) Prostaglandin E2 mediates cellular effects of interleukin-1β on parvocellular neurones in the paraventricular nucleus of the hypothalamus. J Neuroendocrinol 17:498–508. https://doi.org/10.1111/j.1365-2826.2005.01336.x
Florido A, Velasco ER, Monari S et al (2023) Glucocorticoid-based pharmacotherapies preventing PTSD. Neuropharmacology 224:109344. https://doi.org/10.1016/j.neuropharm.2022.109344
Fonkoue IT, Marvar PJ, Norrholm S et al (2020) Symptom severity impacts sympathetic dysregulation and inflammation in post-traumatic stress disorder (PTSD). Brain Behav Immun 83:260–269. https://doi.org/10.1016/j.bbi.2019.10.021
Furuyashiki T, Narumiya S (2011) Stress responses: the contribution of prostaglandin E(2) and its receptors. Nat Rev Endocrinol 7:163–175
Gafford GM, Guo JD, Flandreau EI et al (2012) Cell-type specific deletion of GABA(A) α1 in corticotropin-releasing factor-containing neurons enhances anxiety and disrupts fear extinction. Proc Natl Acad Sci U S A 109:16330–16335. https://doi.org/10.1073/pnas.1119261109
Gazarini L, Jark Stern CA, Carobrez AP, Bertoglio LJ (2013) Enhanced noradrenergic activity potentiates fear memory consolidation and reconsolidation by differentially recruiting α1-and β-adrenergic receptors. Learn Memory 20:210–219. https://doi.org/10.1101/lm.030007.112
Geracioti TD, Baker DG, Ekhator NN et al (2001)CSF Norepinephrine Concentrations in Posttraumatic Stress Disorder
Gopaldas M, Zanderigo F, Zhan S et al (2019) Brain serotonin transporter binding, plasma arachidonic acid and depression severity: a positron emission tomography study of major depression. J Affect Disord 257:495–503. https://doi.org/10.1016/j.jad.2019.07.035
Grah M, Mihanović M, Svrdlin P et al (2010) Serotonin and cortisol as suicidogenic factors in patients with PTSD. Coll Antropol 34:1433–1439
Grinchii D, Dremencov E (2020) Mechanism of action of atypical antipsychotic drugs in mood disorders. Int J Mol Sci 21:1–15. https://doi.org/10.3390/ijms21249532
Groer MW, Kane B, Williams SN, Duffy A (2015) Relationship of PTSD symptoms with Combat exposure, stress, and inflammation in american soldiers. Biol Res Nurs 17:303–310. https://doi.org/10.1177/1099800414544949
Haiju Z, Ruopeng S, Gefei L et al (2009) Cyclooxygenase-2 inhibitor inhibits the hippocampal synaptic reorganization by inhibiting MAPK/ERK activity and modulating GABAergic transmission in pilocarpine-induced status epilepticus rats. Med Chem Res 18:71–90. https://doi.org/10.1007/s00044-008-9109-0
Harnett NG, Wood KH, Ference EW et al (2017) Glutamate/glutamine concentrations in the dorsal anterior cingulate vary with post-traumatic stress disorder symptoms. J Psychiatr Res 91:169–176. https://doi.org/10.1016/j.jpsychires.2017.04.010
Harvey BH, Naciti C, Brand L, Stein DJ (2003) Changes evoked by a time-dependent sensitisation (TDS) stress model in rats. Brain Res 983:97–107
Herman JP, Tasker JG, Ziegler DR, Cullinan WE (2002) Local circuit regulation of paraventricular nucleus stress integration. Pharmacol Biochem Behav 71:457–468. https://doi.org/10.1016/S0091-3057(01)00681-5
Herman JP, Mueller NK, Figueiredo H (2004) Role of GABA and glutamate circuitry in hypothalamo-pituitary- adrenocortical stress integration. Ann N Y Acad Sci 1018:35–45. https://doi.org/10.1196/annals.1296.004
Herr N, Bode C, Duerschmied D (2017) The Effects of Serotonin in Immune cells. Front Cardiovasc Med 4:1–11. https://doi.org/10.3389/fcvm.2017.00048
Holbrook JA, Jarosz-Griffiths HH, Caseley E et al (2021) Neurodegenerative disease and the NLRP3 inflammasome. Front Pharmacol 12:1–15. https://doi.org/10.3389/fphar.2021.643254
Holmes SE, Girgenti MJ, Davis MT et al (2017) Altered metabotropic glutamate receptor 5 markers in PTSD: in vivo and postmortem evidence. Proc Natl Acad Sci 114:8390–8395. https://doi.org/10.1073/pnas.1701749114
Hori H, Kim Y (2019) Inflammation and post-traumatic stress disorder. Psychiatry Clin Neurosci 73:143–153. https://doi.org/10.1111/pcn.12820
Horstmann JP, Marzi I, Relja B (2016) Adrenergic stimulation alters the expression of inflammasome components and interleukins in primary human monocytes. Exp Ther Med 11:297–302. https://doi.org/10.3892/etm.2015.2850
Hoskin JL, Al-Hasan Y, Sabbagh MN (2019) Nicotinic acetylcholine receptor agonists for the treatment of Alzheimer’s dementia: an update. Nicotine and Tobacco Research 21:370–376. https://doi.org/10.1093/ntr/nty116
Howes OD, McCutcheon R, Owen MJ, Murray RM (2017) The role of genes, stress, and dopamine in the development of Schizophrenia. Biol Psychiatry 81:9–20. https://doi.org/10.1016/j.biopsych.2016.07.014
Hung YL, Wang SC, Suzuki K et al (2019) Bavachin attenuates LPS-induced inflammatory response and inhibits the activation of NLRP3 inflammasome in macrophages. Phytomedicine 59:152785. https://doi.org/10.1016/j.phymed.2018.12.008
Hupalo S, Bryce CA, Bangasser DA et al (2019) Corticotropin-releasing factor (CRF) circuit modulation of cognition and motivation. Neurosci Biobehav Rev 103:50–59. https://doi.org/10.1016/j.neubiorev.2019.06.010
Illiano P, Leo D, Gainetdinov RR, Pardo M (2021) Early adolescence prefrontal cortex alterations in female rats lacking dopamine transporter. Biomedicines 9:1–16. https://doi.org/10.3390/biomedicines9020157
Imperato A, Puglisi-Allegra S, Casolini P et al (1989) Stress-induced enhancement of dopamine and acetylcholine release in limbic structures: role of corticosterone. Eur J Pharmacol 165:337–338. https://doi.org/10.1016/0014-2999(89)90735-8
Iwata M, Ota KT, Duman RS (2013) The inflammasome: pathways linking psychological stress, depression, and systemic illnesses. Brain Behav Immun 31:105–114. https://doi.org/10.1016/j.bbi.2012.12.008
Jacek Dębiec DEA, Bush and JEL (2014) Noradrenergic enhancement of reconsolidation in the Amygdala impairs extinction of conditioned fear in rats – a possible mechanism for the persistence of traumatic Memories in PTSD. Bone 23:1–7. https://doi.org/10.1038/jid.2014.371
Jacobsen JPR, Medvedev IO, Caron MG (2012) The 5-HT deficiency theory of depression: perspectives from a naturalistic 5-HT deficiency model, the tryptophan hydroxylase 2Arg439His knockin mouse. Philosophical Trans Royal Soc B: Biol Sci 367:2444–2459. https://doi.org/10.1098/rstb.2012.0109
Jedema HP, Grace AA (2004) Corticotropin-releasing hormone directly activates noradrenergic neurons of the locus ceruleus recorded in vitro. J Neurosci 24:9703–9713. https://doi.org/10.1523/JNEUROSCI.2830-04.2004
Juárez Olguín H, Calderón Guzmán D, Hernández García E, Barragán Mejía G (2016) The role of dopamine and its dysfunction as a consequence of oxidative stress. Oxid Med Cell Longev 2016:. https://doi.org/10.1155/2016/9730467
Judkins JL, Moore BA, Collette TL et al (2020) Incidence rates of posttraumatic stress disorder over a 17-Year period in active Duty Military Service Members. J Trauma Stress 33:994–1006. https://doi.org/10.1002/jts.22558
Kaluski S, Portillo M, Besnard A et al (2017) Neuroprotective functions for the histone deacetylase SIRT6. Cell Rep 18:3052–3062. https://doi.org/10.1016/j.celrep.2017.03.008
Ke P, Shao BZ, Xu ZQ et al (2017) Activating α7 nicotinic acetylcholine receptor inhibits NLRP3 inflammasome through regulation of β-arrestin-1. CNS Neurosci Ther 23:875–884. https://doi.org/10.1111/cns.12758
Kellis DM, Kaigler KF, Witherspoon E et al (2020) Cholinergic neurotransmission in the basolateral amygdala during cued fear extinction. Neurobiol Stress 13:100279. https://doi.org/10.1016/j.ynstr.2020.100279
Kessler RC, Aguilar-Gaxiola S, Alonso J et al (2017) Trauma and PTSD in the WHO World Mental Health surveys. Eur J Psychotraumatol 8. https://doi.org/10.1080/20008198.2017.1353383
Khazaeipool Z, Wiederman M, Inoue W (2018) Prostaglandin E2 depresses GABA release onto parvocellular neuroendocrine neurones in the paraventricular nucleus of the hypothalamus via presynaptic receptors. J Neuroendocrinol 30:0–2. https://doi.org/10.1111/jne.12638
Kim W, Bin, Cho J-H (2020) Encoding of contextual fear memory in hippocampal–amygdala circuit. Nat Commun 11:1382. https://doi.org/10.1038/s41467-020-15121-2
Kim H, Kim HS, Kaang BK (2018) Elevated contextual fear memory by SIRT6 depletion in excitatory neurons of mouse forebrain. Mol Brain 11. https://doi.org/10.1186/s13041-018-0391-6
Kinra M, Joseph A, Nampoothiri M et al (2021) Inhibition of NLRP3-inflammasome mediated IL-1β release by phenylpropanoic acid derivatives: in-silico and in-vitro approach. Eur J Pharm Sci 157:105637. https://doi.org/10.1016/j.ejps.2020.105637
Kita I, Seki Y, Nakatani Y et al (2006) Corticotropin-releasing factor neurons in the hypothalamic paraventricular nucleus are involved in arousal/yawning response of rats. Behav Brain Res 169:48–56. https://doi.org/10.1016/j.bbr.2005.12.003
Klein MO, Battagello DS, Cardoso AR et al (2019) Dopamine: functions, signaling, and Association with neurological Diseases. Cell Mol Neurobiol 39:31–59. https://doi.org/10.1007/s10571-018-0632-3
Koirala R, Aass HCD, Søegaard EGI et al (2023) Association of pro-inflammatory cytokines with trauma and post-traumatic stress disorder visiting a tertiary care hospital in Kathmandu. PLoS ONE 18:e0281125. https://doi.org/10.1371/journal.pone.0281125
Kolko M, DeCoster MA, de Turco EBR, Bazan NG (1996) Synergy by secretory phospholipase A2 and glutamate on inducing cell death and sustained arachidonic acid metabolic changes in primary cortical neuronal cultures. J Biol Chem 271:32722–32728. https://doi.org/10.1074/jbc.271.51.32722
Kolko M, Nielsen M, Bazan NG, Diemer NH (2002) Secretory phospholipase A2 induces delayed neuronal COX-2 expression compared with glutamate. J Neurosci Res 69:169–177. https://doi.org/10.1002/jnr.10288
Kurrasch-Orbaugh DM, Parrish JC, Watts VJ, Nichols DE (2003) A complex signaling cascade links the serotonin2A receptor to phospholipase A2 activation: the involvement of MAP kinases. J Neurochem 86:980–991. https://doi.org/10.1046/j.1471-4159.2003.01921.x
Lanzenberger R, Wadsak W, Spindelegger C et al (2010) Cortisol plasma levels in social anxiety disorder patients correlate with serotonin-1A receptor binding in limbic brain regions. Int J Neuropsychopharmacol 13:1129–1143. https://doi.org/10.1017/S1461145710000581
Lappas M (2012) Anti-inflammatory properties of sirtuin 6 in human umbilical vein endothelial cells. Mediators Inflamm 2012. https://doi.org/10.1155/2012/597514
Lawford BR, Young R, Noble EP et al (2006) The D2 dopamine receptor (DRD2) gene is associated with co-morbid depression, anxiety and social dysfunction in untreated veterans with post-traumatic stress disorder. Eur Psychiatry 21:180–185. https://doi.org/10.1016/j.eurpsy.2005.01.006
Lee B, Sur B, Lee H, Oh S (2020) Korean Red Ginseng prevents posttraumatic stress disorder–triggered depression-like behaviors in rats via activation of the serotonergic system. J Ginseng Res 44:644–654. https://doi.org/10.1016/j.jgr.2019.09.005
Li CT, Yang KC, Lin WC (2019a) Glutamatergic dysfunction and glutamatergic compounds for major psychiatric disorders: evidence from clinical neuroimaging studies. Front Psychiatry 10:1–11. https://doi.org/10.3389/fpsyt.2018.00767
Li L, Liu Z, Jiang YY et al (2019b) Acetylcholine suppresses microglial inflammatory response via α7nAChR to protect hippocampal neurons. J Integr Neurosci 18:51–56. https://doi.org/10.31083/j.jin.2019.01.114
Li W, Guo B, Tao K et al (2019c) Inhibition of SIRT1 in hippocampal CA1 ameliorates PTSD-like behaviors in mice by protections of neuronal plasticity and serotonin homeostasis via NHLH2/MAO-A pathway. Biochem Biophys Res Commun 518:344–350. https://doi.org/10.1016/j.bbrc.2019.08.060
Libert S, Pointer K, Bell EL et al (2011) SIRT1 activates MAO-A in the brain to mediate anxiety and exploratory drive. Cell 147:1459–1472. https://doi.org/10.1016/j.cell.2011.10.054
Lin S, Mei X (2021) Role of NLRP3 inflammasomes in Neuroinflammation Diseases. Eur Neurol 83:576–580. https://doi.org/10.1159/000509798
Liu L, Xia G, Li P et al (2021) Sirt-1 regulates physiological process and exerts protective effects against oxidative stress. Biomed Res Int 2021. https://doi.org/10.1155/2021/5542545
Luo FF, Han F, Shi YX (2011) Changes in 5-HT1A receptor in the dorsal raphe nucleus in a rat model of post-traumatic stress disorder. Mol Med Rep 4:843–847. https://doi.org/10.3892/mmr.2011.516
Mackowiak M, Chocyk A, Sanak M et al (2002) DOI, an agonist of 5-HT2A/2 C serotonin receptor, alters the expression of cyclooxygenase-2 in the rat parietal cortex. J Physiol Pharmacol 53:395–407
Markus A, John AB, Chadi HK (2016) Glutamate dysregulation and glutamatergic therapeutics for PTSD: evidence from Human Studies. Neurosci Lett 649:147–155. https://doi.org/10.1016/j.neulet.2016.11.064
Mather M, Clewett D, Sakaki M, Harley CW (2016) Norepinephrine ignites local hotspots of neuronal excitation: how arousal amplifies selectivity in perception and memory. Behav Brain Sci 39. https://doi.org/10.1017/S0140525X15000667
Mellman TA, Alim T, Brown DD et al (2009) Serotonin polymorphisms and posttraumatic stress disorder in a trauma exposed african american population. Depress Anxiety 26:993–997. https://doi.org/10.1002/da.20627
Mellon SH, Gautam A, Hammamieh R et al (2018) Metabolism, Metabolomics, and inflammation in posttraumatic stress disorder. Biol Psychiatry 83:866–875. https://doi.org/10.1016/j.biopsych.2018.02.007
Mendoza C, Barreto GE, Iarkov A et al (2018) Cotinine: a therapy for memory extinction in post-traumatic stress disorder. Mol Neurobiol 55:6700–6711. https://doi.org/10.1007/s12035-018-0869-3
Meyerhoff DJ, Mon A, Metzler T, Neylan TC (2014) Cortical gamma-aminobutyric acid and glutamate in posttraumatic stress disorder and their relationships to self-reported sleep quality. Sleep 37:893–900. https://doi.org/10.5665/sleep.3654
Mizoguchi K, Yuzurihara M, Ishige A et al (2000) Chronic stress induces impairment of spatial working memory because of prefrontal dopaminergic dysfunction. J Neurosci 20:1568–1574. https://doi.org/10.1523/jneurosci.20-04-01568.2000
Mody I, Maguire J (2012) The reciprocal regulation of stress hormones and GABA A receptors. Front Cell Neurosci 6:1–6. https://doi.org/10.3389/fncel.2012.00004
Moore BA, Pujol L, Waltman S, Shearer DS (2021) Management of post-traumatic stress disorder in Veterans and Military Service Members: a review of pharmacologic and psychotherapeutic interventions since 2016. Curr Psychiatry Rep 23. https://doi.org/10.1007/s11920-020-01220-w
Morena M, Patel S, Bains JS, Hill MN (2016) Neurobiological interactions between stress and the Endocannabinoid System. Neuropsychopharmacology 41:80–102. https://doi.org/10.1038/npp.2015.166
Murnane KS (2019) Serotonin 2A receptors are a stress response system: implications for post-traumatic stress disorder. Behav Pharmacol 30:151–162. https://doi.org/10.1097/FBP.0000000000000459
Myers B, Mark Dolgas C, Kasckow J et al (2014) Central stress-integrative circuits: Forebrain glutamatergic and GABAergic projections to the dorsomedial hypothalamus, medial preoptic area, and bed nucleus of the stria terminalis. Brain Struct Funct 219:1287–1303. https://doi.org/10.1007/s00429-013-0566-y
Naegeli C, Zeffiro T, Piccirelli M et al (2018) Locus Coeruleus Activity mediates hyperresponsiveness in posttraumatic stress disorder. Biol Psychiatry 83:254–262. https://doi.org/10.1016/j.biopsych.2017.08.021
Nasca C, Orlando R, Marchiafava M et al (2013) Exposure to predator odor and resulting anxiety enhances the expression of the α2δ subunit of voltage-sensitive calcium channels in the amygdala. J Neurochem 125:649–656. https://doi.org/10.1111/j.1471-4159.2012.07895.x
Nasir M, Trujillo D, Levine J et al (2020) Glutamate Systems in DSM-5 anxiety Disorders: their role and a review of glutamate and GABA psychopharmacology. https://doi.org/10.3389/fpsyt.2020.548505. Front Psychiatry 11:
Nautiyal KM, Tritschler L, Ahmari SE et al (2016) A lack of serotonin 1B autoreceptors results in decreased anxiety and depression-related behaviors. Neuropsychopharmacology 41:2941–2950. https://doi.org/10.1038/npp.2016.109
Navarro E, Norden DM, Trojanowski PJ et al (2021) Central activation of alpha7 nicotinic signaling attenuates LPS-induced neuroinflammation and sickness behavior in adult but not in aged animals. Molecules 26. https://doi.org/10.3390/molecules26082107
Neumaier JF, Sexton TJ, Hamblin MW, Beck SG (2000) Corticosteroids regulate 5-HT1A but not 5-HT1B receptor mRNA in rat hippocampus. Mol Brain Res 82:65–73. https://doi.org/10.1016/S0169-328X(00)00181-9
Nicholson EL, Bryant RA, Felmingham KL (2014) Interaction of noradrenaline and cortisol predicts negative intrusive memories in posttraumatic stress disorder. Neurobiol Learn Mem 112:204–211. https://doi.org/10.1016/j.nlm.2013.11.018
Nie X, Kitaoka S, Shinohara M et al (2019) Roles of toll-like receptor 2/4, monoacylglycerol lipase, and cyclooxygenase in social defeat stress-induced prostaglandin E2 synthesis in the brain and their behavioral relevance. Sci Rep 9:1–10. https://doi.org/10.1038/s41598-019-54082-5
Nisar S, Bhat AA, Hashem S et al (2020) Genetic and neuroimaging approaches to understanding post-traumatic stress disorder. Int J Mol Sci 21:1–21. https://doi.org/10.3390/ijms21124503
Nishi D, Hashimoto K, Noguchi H et al (2015) Glutamatergic system abnormalities in posttraumatic stress disorder. Psychopharmacology 232:4261–4268. https://doi.org/10.1007/s00213-015-4052-5
Northrop NA, Yamamoto BK (2013) Cyclooxygenase activity contributes to the monoaminergic damage caused by serial exposure to stress and methamphetamine. Neuropharmacology 72:96–105. https://doi.org/10.1016/j.neuropharm.2013.04.040
Nwokafor C, Serova LI, Tanelian A et al (2021) Variable response of Norepinephrine Transporter to traumatic stress and relationship to Hyperarousal. Front Behav Neurosci 15. https://doi.org/10.3389/fnbeh.2021.725091
O’Donnell T, Hegadoren KM, Coupland NC (2004) Noradrenergic mechanisms in the pathophysiology of post-traumatic stress disorder. Neuropsychobiology 50:273–283. https://doi.org/10.1159/000080952
O’Toole BI, Gorman P, Catts S (2020) Military combat, posttraumatic stress disorder, and the course of Alcohol Use Disorders in a cohort of australian Vietnam War Veterans. J Trauma Stress 33:709–719. https://doi.org/10.1002/jts.22588
Olson VG, Rockett HR, Reh RK et al (2011) The role of norepinephrine in differential response to stress in an animal model of posttraumatic stress disorder. Biol Psychiatry 70:441–448. https://doi.org/10.1016/j.biopsych.2010.11.029
Otsuka T, Hori H, Yoshida F et al (2021) Association of CRP genetic variation with symptomatology, cognitive function, and circulating proinflammatory markers in civilian women with PTSD. J Affect Disord 279:640–649. https://doi.org/10.1016/j.jad.2020.10.045
Parsadaniantz SM, Lebeau A, Duval P et al (2000) Effects of the inhibition of cyclo-oxygenase 1 or 2 or 5-lipoxygenase on the activation of the hypothalamic-pituitary-adrenal axis induced by interleukin-1 β in the male rat. J Neuroendocrinol 12:766–773. https://doi.org/10.1046/j.1365-2826.2000.00517.x
Patel H, McIntire J, Ryan S et al (2017) Anti-inflammatory effects of astroglial α7 nicotinic acetylcholine receptors are mediated by inhibition of the NF-ΚB pathway and activation of the Nrf2 pathway. J Neuroinflammation 14:1–15. https://doi.org/10.1186/s12974-017-0967-6
Paul S, Jeon WK, Bizon JL, Han JS (2015) Interaction of basal forebrain cholinergic neurons with the glucocorticoid system in stress regulation and cognitive impairment. Front Aging Neurosci 7:1–11. https://doi.org/10.3389/fnagi.2015.00043
Pavlovsky L, Bitan Y, Shalev H et al (2012) Stress-induced altered cholinergic-glutamatergic interactions in the mouse hippocampus. Brain Res 1472:99–106. https://doi.org/10.1016/j.brainres.2012.05.057
Peng-Fei H, A-Ru-Na, Hui C et al (2021) Activation of alpha7 nicotinic acetylcholine receptor protects bovine endometrial tissue against LPS-induced inflammatory injury via JAK2/STAT3 pathway and COX-2 derived prostaglandin E2. Eur J Pharmacol 900:174067. https://doi.org/10.1016/j.ejphar.2021.174067
Pervanidou P, GP chrousos (2012) Posttraumatic stress disorder in children and adolescents. Sci signal. https://doi.org/10.1007/978-1-4614-9608-3_38. 5:
Pietrzak RH, Gallezot JD, Ding YS et al (2013a) Association of posttraumatic stress disorder with reduced in vivo norepinephrine transporter availability in the locus coeruleus. JAMA Psychiatry 70:1199–1205. https://doi.org/10.1001/jamapsychiatry.2013.399
Pietrzak RH, Henry S, Southwick SM et al (2013b) Linking in vivo brain serotonin type 1B receptor density to phenotypic heterogeneity of posttraumatic stress symptomatology. Mol Psychiatry 18:399–401. https://doi.org/10.1038/mp.2012.60
Pillai L, Mehta SG, Chaudhari BL (2016) Post-traumatic stress disorder (PTSD): indian perspective. Comprehensive Guide to post-traumatic stress Disorders. Springer International Publishing, Cham, pp 1617–1635
Piovesana R, Intriago MSS, Dini L, Tata AM (2021) Cholinergic modulation of neuroinflammation: focus on α7 nicotinic receptor. Int J Mol Sci 22. https://doi.org/10.3390/ijms22094912
Polter AM, Li X (2010) 5-HT1A receptor-regulated signal transduction pathways in brain. Cell Signal 22:1406–1412. https://doi.org/10.1016/j.cellsig.2010.03.019
Prajapati SK, Krishnamurthy S (2021) Development and treatment of cognitive inflexibility in sub-chronic stress–re-stress (SRS) model of PTSD. Pharmacol Rep 73:464–479. https://doi.org/10.1007/s43440-020-00198-9
Quan N, He L, Lai W (2003) Endothelial activation is an intermediate step for peripheral lipopolysaccharide induced activation of paraventricular nucleus. Brain Res Bull 59:447–452. https://doi.org/10.1016/S0361-9230(02)00951-6
Rajbhandari AK, Bakshi VP (2020) Repeated norepinephrine receptor stimulation in the BNST induces sensorimotor gating deficits via corticotropin releasing factor. Neuropharmacology 172:108090. https://doi.org/10.1016/j.neuropharm.2020.108090
Ranadive N, Arora D, Nampoothiri M, Mudgal J (2021) Sirtuins, a potential target in traumatic Brain Injury and relevant experimental models. Brain Res Bull 171:135–141
Règue M, Poilbout C, Martin V et al (2019) Increased 5-HT2C receptor editing predisposes to PTSD-like behaviors and alters BDNF and cytokines signaling. Transl Psychiatry 9. https://doi.org/10.1038/s41398-019-0431-8
Reul JMHM, Nutt DJ (2008) Glutamate and cortisol - A critical confluence in PTSD? J Psychopharmacol 22:469–472. https://doi.org/10.1177/0269881108094617
Rice O, Ashby CR, Dixon C et al (2018) Selective dopamine D 3 receptor antagonism significantly attenuates stress-induced immobility in a rat model of post-traumatic stress disorder. Synapse 72. https://doi.org/10.1002/syn.22035
Rohleder N, Joksimovic L, Wolf JM, Kirschbaum C (2004) Hypocortisolism and increased glucocorticoid sensitivity of pro-inflammatory cytokine production in bosnian war refugees with posttraumatic stress disorder. Biol Psychiatry 55:745–751. https://doi.org/10.1016/j.biopsych.2003.11.018
Ronzoni G, del Arco A, Mora F, Segovia G (2016) Enhanced noradrenergic activity in the amygdala contributes to hyperarousal in an animal model of PTSD. Psychoneuroendocrinology 70:1–9. https://doi.org/10.1016/j.psyneuen.2016.04.018
Rosso IM, Crowley DJ, Silveri MM et al (2017) Hippocampus glutamate and N-Acetyl aspartate markers of excitotoxic neuronal compromise in posttraumatic stress disorder. Neuropsychopharmacology 42:1698–1705. https://doi.org/10.1038/npp.2017.32
Sabban EL, Serova LI, Newman E et al (2018) Changes in Gene expression in the Locus Coeruleus-Amygdala Circuitry in Inhibitory Avoidance PTSD Model. Cell Mol Neurobiol 38:273–280. https://doi.org/10.1007/s10571-017-0548-3
Sarkar P, Mozumder S, Bej A et al (2021) Structure, dynamics and lipid interactions of serotonin receptors: excitements and challenges. Biophys Rev 13:101–122. https://doi.org/10.1007/s12551-020-00772-8
Sauvage M, Steckler T (2001) Detection of corticotropin-releasing hormone receptor 1 immunoreactivity in cholinergic, dopaminergic and noradrenergic neurons of the murine basal forebrain and brainstem nuclei - potential implication for arousal and attention. Neuroscience 104:643–652. https://doi.org/10.1016/S0306-4522(01)00137-3
Shajib MS, Khan WI (2015) The role of serotonin and its receptors in activation of immune responses and inflammation. Acta Physiol 213:561–574. https://doi.org/10.1111/apha.12430
Sharma D, Farrar JD (2020) Adrenergic regulation of immune cell function and inflammation. Semin Immunopathol 42:709–717. https://doi.org/10.1007/s00281-020-00829-6
Sherin JE, Nemeroff CB (2011) Post-traumatic stress disorder: the neurobiological impact of psychological trauma. Dialogues Clin Neurosci 13:263–278. https://doi.org/10.31887/dcns.2011.13.2/jsherin
Sheth C, Prescot AP, Legarreta M et al (2019) Reduced gamma-amino butyric acid (GABA) and glutamine in the anterior cingulate cortex (ACC) of veterans exposed to trauma. J Affect Disord 248:166–174. https://doi.org/10.1016/j.jad.2019.01.037
Shimizu T, Tanaka K, Nakamura K et al (2014) Possible involvement of brain prostaglandin E2 and prostanoid EP3 receptors in prostaglandin E2 glycerol ester-induced activation of central sympathetic outflow in the rat. Neuropharmacology 82:19–27. https://doi.org/10.1016/j.neuropharm.2014.03.005
Song D, Ge Y, Chen Z et al (2018) Role of dopamine D3 receptor in alleviating behavioural deficits in animal models of post-traumatic stress disorder. Prog Neuropsychopharmacol Biol Psychiatry 84:190–200. https://doi.org/10.1016/j.pnpbp.2018.03.001
Srikumar BN, Raju TR, Shankaranarayana Rao BS (2006) The involvement of cholinergic and noradrenergic systems in behavioral recovery following oxotremorine treatment to chronically stressed rats. Neuroscience 143:679–688. https://doi.org/10.1016/j.neuroscience.2006.08.041
Stephens MAC, Wand G (2012) Stress and the HPA axis: role of glucocorticoids in alcohol dependence. Alcohol Res 34:468–483. https://doi.org/10.1016/B978-0-12-405941-2.00021-3
Strosznajder J, Samochocki M, Duran M (1994) Serotonin, a potent modulator of arachidonic acid turnover, interaction with glutamatergic receptor in brain cortex. Neurochem Int 25:193–199. https://doi.org/10.1016/0197-0186(94)90039-6
Sukhmanjeet Kaur Mann; Raman Marwaha (2021) Posttraumatic Stress Disorder. In: StatPearls. https://www.ncbi.nlm.nih.gov/books/NBK559129/
Sullivan GM, Ogden RT, Huang YY et al (2013) Higher in vivo serotonin-1A binding in posttraumatic stress disorder: a pet study with [11 C]WAY-100635. Depress Anxiety 30:197–206. https://doi.org/10.1002/da.22019
Sun R, Zhang W, Bo J et al (2017) Spinal activation of alpha7-nicotinic acetylcholine receptor attenuates posttraumatic stress disorder-related chronic pain via suppression of glial activation. Neuroscience 344:243–254. https://doi.org/10.1016/j.neuroscience.2016.12.029
Swanson K, Deng M, Ting JPY (2019) The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 19:477–489. https://doi.org/10.1038/s41577-019-0165-0
Tafet GE, Nemeroff CB (2016) The links between stress and depression: Psychoneuroendocrinological, genetic, and environmental interactions. J Neuropsychiatry Clin Neurosci 28:77–88. https://doi.org/10.1176/appi.neuropsych.15030053
Tan KS, Nackley AG, Satterfield K et al (2007) β2 adrenergic receptor activation stimulates pro-inflammatory cytokine production in macrophages via PKA- and NF-κB-independent mechanisms. Cell Signal 19:251–260. https://doi.org/10.1016/j.cellsig.2006.06.007
Tiwari P, Dwivedi S, Singh MP et al (2013) Basic and modern concepts on cholinergic receptor: a review. Asian Pac J Trop Dis 3:413–420. https://doi.org/10.1016/S2222-1808(13)60094-8
Toorie AM, Cyr NE, Steger JS et al (2016) The nutrient and energy sensor Sirt1 regulates the hypothalamic-pituitary-adrenal (HPA) axis by altering the production of the Prohormone Convertase 2 (PC2) essential in the maturation of corticotropin-releasing hormone (CRH) from its prohormone in male rats. J Biol Chem 291:5844–5859. https://doi.org/10.1074/jbc.M115.675264
Torrisi SA, Leggio GM, Drago F, Salomone S (2019) Therapeutic challenges of posttraumatic stress disorder: focus on the dopaminergic system. Front Pharmacol 10:1–11. https://doi.org/10.3389/fphar.2019.00404
Trousselard M, Lefebvre B, Andruetan Y et al (2016) Is plasma GABA level a biomarker of Post- traumatic stress disorder (PTSD) severity? A preliminary study. Psychiatry Res 241:273–2279. https://doi.org/10.1016/j.psychres.2016.05.013
Tung S, Hardy AB, Wheeler MB, Belsham DD (2012) Serotonin (5-HT) activation of immortalized hypothalamic neuronal cells through the 5-HT1Bserotonin receptor. Endocrinology 153:4862–4873. https://doi.org/10.1210/en.2012-1538
Tyagi E, Agrawal R, Nath C, Shukla R (2010) Cholinergic protection via α7 nicotinic acetylcholine receptors and PI3K-Akt pathway in LPS-induced neuroinflammation. Neurochem Int 56:135–142. https://doi.org/10.1016/j.neuint.2009.09.011
Ulmer CS, Hall MH, Dennis PA et al (2018) Posttraumatic stress disorder diagnosis is associated with reduced parasympathetic activity during sleep in US veterans and military service members of the Iraq and Afghanistan wars. Sleep 41:1–9. https://doi.org/10.1093/sleep/zsy174
Uniyal A, Singh R, Akhtar A et al (2019) Co-treatment of piracetam with risperidone rescued extinction deficits in experimental paradigms of post-traumatic stress disorder by restoring the physiological alterations in cortex and hippocampus. Pharmacol Biochem Behav 185:172763. https://doi.org/10.1016/j.pbb.2019.172763
Vidal PM, Pacheco R (2020) The Cross-Talk between the dopaminergic and the Immune System involved in Schizophrenia. Front Pharmacol 11:1–18. https://doi.org/10.3389/fphar.2020.00394
Walker SW, Strachan MWJ, Lightly ERT et al (1990) Acetylcholine stimulates cortisol secretion through the M3 muscarinic receptor linked to a polyphosphoinositide-specific phospholipase C in bovine adrenal fasciculata/reticularis cells. Mol Cell Endocrinol 72:227–238. https://doi.org/10.1016/0303-7207(90)90147-Z
Wang M, Duan F, Wu J et al (2018a) Effect of cyclooxygenase-2 inhibition on the development of post-traumatic stress disorder in rats. Mol Med Rep 17:4925–4932. https://doi.org/10.3892/mmr.2018.8525
Wang T, Nowrangi D, Yu L et al (2018b) Activation of dopamine D1 receptor decreased NLRP3-mediated inflammation in intracerebral hemorrhage mice. J Neuroinflammation 15:1–10. https://doi.org/10.1186/s12974-017-1039-7
Wang J, Wang F, Mai D, Qu S (2020) Molecular Mechanisms of Glutamate Toxicity in Parkinson’s Disease. Front Neurosci 14:1–12. https://doi.org/10.3389/fnins.2020.585584
Watanabe M, Maemura K, Kanbara K et al (2002) GABA and GABA receptors in the central nervous system and other organs. Int Rev Cytol 213:1–47. https://doi.org/10.1016/S0074-7696(02)13011-7
Watson P (2019) PTSD as a public Mental Health Priority. Curr Psychiatry Rep 21. https://doi.org/10.1007/s11920-019-1032-1
Weathers FW, Marx BP, Friedman MJ, Schnurr PP (2014) Posttraumatic stress disorder in DSM-5: New Criteria, New Measures, and implications for Assessment. Psychol Inj Law 7:93–107. https://doi.org/10.1007/s12207-014-9191-1
Wei P, Yang F, Zheng Q et al (2019) The potential role of the NLRP3 inflammasome activation as a link between mitochondria ROS generation and neuroinflammation in postoperative cognitive dysfunction. Front Cell Neurosci 13:1–10. https://doi.org/10.3389/fncel.2019.00073
Whitney MS, Shemery AM, Yaw AM et al (2016) Adult brain serotonin deficiency causes hyperactivity, circadian disruption, and elimination of siestas. J Neurosci 36:9828–9842. https://doi.org/10.1523/JNEUROSCI.1469-16.2016
Winek K, Soreq H, Meisel A (2021) Regulators of cholinergic signaling in disorders of the central nervous system. J Neurochem 1–14. https://doi.org/10.1111/jnc.15332
Wingenfeld K, Whooley MA, Neylan TC et al (2015) Effect of current and lifetime posttraumatic stress disorder on 24-h urinary catecholamines and cortisol: results from the mind your heart study. Psychoneuroendocrinology 52:83–91. https://doi.org/10.1016/j.psyneuen.2014.10.023
Wu ZM, Zheng CH, Zhu ZH et al (2016) SiRNA-mediated serotonin transporter knockdown in the dorsal raphe nucleus rescues single prolonged stress-induced hippocampal autophagy in rats. J Neurol Sci 360:133–140. https://doi.org/10.1016/j.jns.2015.11.056
Wu H, Denna TH, Storkersen JN, Gerriets VA (2019) Beyond a neurotransmitter: the role of serotonin in inflammation and immunity. Pharmacol Res 140:100–114. https://doi.org/10.1016/j.phrs.2018.06.015
Xia Y, He F, Wu X et al (2021) GABA transporter sustains IL-1β production in macrophages. Sci Adv 7. https://doi.org/10.1126/SCIADV.ABE9274
Yamaguchi N, Okada S (2009) Cyclooxygenase-1 and – 2 in spinally projecting neurons are involved in CRF-induced sympathetic activation. Auton Neurosci 151:82–89. https://doi.org/10.1016/j.autneu.2009.06.009
Yamamoto M, Takahashi Y (2018) The essential role of sirt1 in hypothalamic-pituitary axis.Front Endocrinol (Lausanne)9
Yamanashi T, Iwata M, Shibushita M et al (2020) Beta-hydroxybutyrate, an endogenous NLRP3 inflammasome inhibitor, attenuates anxiety-related behavior in a rodent post-traumatic stress disorder model. Sci Rep 10:1–11. https://doi.org/10.1038/s41598-020-78410-2
Yan Y, Jiang W, Liu L et al (2015) Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 160:62–73. https://doi.org/10.1016/j.cell.2014.11.047
Yang S-J, Han AR, Kim E et al (2019a) KHG21834 attenuates glutamate-induced mitochondrial damage, apoptosis, and NLRP3 inflammasome activation in SH-SY5Y human neuroblastoma cells. Eur J Pharmacol 856:172412. https://doi.org/10.1016/j.ejphar.2019.172412
Yang Y, Wang H, Kouadir M et al (2019b) Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis 10. https://doi.org/10.1038/s41419-019-1413-8
Yang R, Gautam A, Getnet D et al (2020) Epigenetic biotypes of post-traumatic stress disorder in war-zone exposed veteran and active duty males. Mol Psychiatry. https://doi.org/10.1038/s41380-020-00966-2
Yanpallewar S, Tomassoni-Ardori F, Palko ME et al (2022) TrkA-cholinergic signaling modulates fear encoding and extinction learning in PTSD-like behavior. Transl Psychiatry 12. https://doi.org/10.1038/s41398-022-01869-2
Yeong KY, Berdigaliyev N, Chang Y (2020) Sirtuins and their implications in neurodegenerative Diseases from a drug Discovery Perspective. ACS Chem Neurosci 11:4073–4091. https://doi.org/10.1021/acschemneuro.0c00696
Yin X, Gao Y, Shi HS et al (2016) Overexpression of SIRT6 in the hippocampal CA1 impairs the formation of long-term contextual fear memory. Sci Rep 6:1–11. https://doi.org/10.1038/srep18982
Yohn CN, Gergues MM, Samuels BA (2017) The role of 5-HT receptors in depression. Mol Brain 10:1–12. https://doi.org/10.1186/s13041-017-0306-y
Yokotani K, Wang M, Murakami Y et al (2000) Brain phospholipase a 2 -arachidonic acid cascade is involved in the activation of central sympatho-adrenomedullary outflow in rats.341–347
Yuan M, Zhu H, Li Y et al (2021) The DRD2 Taq1A polymorphism moderates the effect of PTSD symptom severity on the left hippocampal CA3 volume: a pilot study. Psychopharmacology. https://doi.org/10.1007/s00213-021-05882-z
Zefferino R, di Gioia S, Conese M (2021) Molecular links between endocrine, nervous and immune system during chronic stress. Brain Behav 11:1–15. https://doi.org/10.1002/brb3.1960
Zhang ZH, Yu Y, Wei SG et al (2011) EP3 receptors mediate PGE2-induced hypothalamic paraventricular nucleus excitation and sympathetic activation. Am J Physiol Heart Circ Physiol 301:1559–1569. https://doi.org/10.1152/ajpheart.00262.2011
Zhang Z, Xu X, Ma J et al (2013) Gene deletion of Gabarap enhances NLRP3 inflammasome-dependent inflammatory responses. J Immunol 190:3517–3524. https://doi.org/10.4049/jimmunol.1202628
Zhang L, Fan Y, Su H et al (2018) Chlorogenic acid methyl ester exerts strong anti-inflammatory effects: Via inhibiting the COX-2/NLRP3/NF-κB pathway. Food Funct 9:6155–6164. https://doi.org/10.1039/c8fo01281d
Zhang L, Hu XZ, Li X et al (2020) Potential chemokine biomarkers associated with PTSD onset, risk and resilience as well as stress responses in US military service members. Transl Psychiatry 10:1–9. https://doi.org/10.1038/s41398-020-0693-1
Zhou Y, Danbolt NC (2014) Glutamate as a neurotransmitter in the healthy brain. J Neural Transm 121:799–817. https://doi.org/10.1007/s00702-014-1180-8
Zhou P, Deng M, Wu J et al (2021) Ventral Tegmental Area Dysfunction and disruption of dopaminergic homeostasis: implications for post-traumatic stress disorder. Mol Neurobiol 58:2423–2434. https://doi.org/10.1007/s12035-020-02278-6
Zhou C, Zheng J, Fan Y, Wu J (2022) TI: NLRP3 inflammasome-dependent pyroptosis in CNS trauma: a potential therapeutic target.Front Cell Dev Biol10
Zuschlag ZD, Compean E, Nietert P et al (2021) Dopamine transporter (DAT1) gene in combat veterans with PTSD: a case-control study. Psychiatry Res 298:113801. https://doi.org/10.1016/j.psychres.2021.113801
Acknowledgements
The authors wish to acknowledge the infrastructural support provided by the Department of Pharmacology, Manipal College of Pharmaceutical Sciences, and Manipal Academy of Higher Education, Manipal-576104, India. The authors also would like to thank the Department of Science and Technology (DST), New Delhi, India for appointing AG under the Women Scientist-A (WOS-A) scheme (DST/WOS-A/LS-37/2021). The authors are grateful to the developers of Inkscape version 1.2 (https://inkscape.org/) free version which was used for the development of the images in the manuscript except Fig. 3 (created by using Microsoft PowerPoint 2019).
Funding
Open access funding provided by Manipal Academy of Higher Education, Manipal
Author information
Authors and Affiliations
Contributions
All the authors contributed to the collection of evidence and the drafting of the manuscript. Anusha Govindula wrote the initial draft of the paper, and also designed the figures [using Inkscape 1.2 (https://inkscape.org/) and Microsoft Powerpoint 2019]. Anusha Govindula and Niraja Ranadive contributed to revising the draft, fact-checking, editing and finalizing the manuscript. Madhavan Nampoothiri and C Mallikarjuna Rao edited and modified the manuscript and provided critical comments. All authors commented and provided feedback on previous versions of the manuscript. Devinder Arora and Jayesh Mudgal contributed equally to conceptualizing, designing and final preparation of this manuscript. The final manuscript was reviewed and approved by all authors.
Corresponding authors
Ethics declarations
Conflicts of interest/Competing Interests
Not applicable.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for publication
Not applicable.
Declarations of Interest
None.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Govindula, A., Ranadive, N., Nampoothiri, M. et al. Emphasizing the Crosstalk Between Inflammatory and Neural Signaling in Post-traumatic Stress Disorder (PTSD). J Neuroimmune Pharmacol 18, 248–266 (2023). https://doi.org/10.1007/s11481-023-10064-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-023-10064-z