
Abstract
Arrhythmic manifestations of COVID-19 include atrial arrhythmias such as atrial fibrillation or atrial flutter, sinus node dysfunction, atrioventricular conduction abnormalities, ventricular tachyarrhythmias, sudden cardiac arrest, and cardiovascular dysautonomias including the so-called long COVID syndrome. Various pathophysiological mechanisms have been implicated, such as direct viral invasion, hypoxemia, local and systemic inflammation, changes in ion channel physiology, immune activation, and autonomic dysregulation. The development of atrial or ventricular arrhythmias in hospitalized COVID-19 patients has been shown to portend a higher risk of in-hospital death. Management of these arrhythmias should be based on published evidence-based guidelines, with special consideration of the acuity of COVID-19 infection, concomitant use of antimicrobial and anti-inflammatory drugs, and the transient nature of some rhythm disorders. In view of new SARS-CoV‑2 variants that may evolve, the development and use of newer antiviral and immunomodulator drugs, and the increasing adoption of vaccination, clinicians must remain vigilant for other arrhythmic manifestations that may occur in association with this novel but potentially deadly disease.
Zusammenfassung
Zu den arrhythmischen Manifestationen von COVID-19 zählen Vorhofarrhythmien wie Vorhofflimmern und Vorhofflattern, Fehlfunktionen des Sinusknotens, atrioventrikuläre Leitungsstörungen, Kammer-Tachyarrhythmien, plötzlicher Herzstillstand und kardiovaskuläre Dysautonomien einschließlich des „Long-COVID-Syndroms“. Verschiedene pathophysiologische Mechanismen wurden damit in Zusammenhang gebracht, wie etwa eine direkte virale Invasion, Hypoxämie, lokale und systemische Entzündung, Änderungen in der Ionenkanal-Physiologie, Immunaktivierung oder autonome Fehlsteuerung. Es hat sich gezeigt, dass Vorhof- oder Kammerarrhythmien bei hospitalisierten COVID-19-Patienten auf eine erhöhte Krankenhaussterblichkeit hinweisen. Die Behandlung dieser Arrhythmien sollte sich an evidenzbasierten Leitlinien orientieren, unter besonderer Berücksichtigung des Ausmaßes der COVID-19-Infektion, des gleichzeitigen Einsatzes von antimikrobiellen und antientzündlichen Medikamenten sowie des vorübergehenden Charakters mancher Rhythmusstörungen. Angesichts der möglichen Entstehung neuer SARS-CoV-2-Varianten, der Entwicklung und des Einsatzes neuer antiviraler und immunmodulatorischer Medikamente sowie der zunehmenden Verwendung von Impfstoffen ist es angezeigt, dass Kliniker aufmerksam auf andere arrhythmische Manifestationen achten, die möglicherweise im Zusammenhang mit dieser neuartigen, potenziell tödlichen Erkrankung auftreten.
Similar content being viewed by others

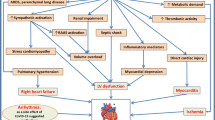
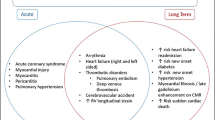
According to the World Health Organization, by 16 March 2023, SARS-CoV‑2 had infected more than 760 million people worldwide, with more than 6.8 million deaths (0.9%). The most common extrapulmonary manifestations of COVID-19 involve the cardiovascular system, and case series early in the evolution of the pandemic reported that cardiac injury (manifested as elevated cardiac biomarker levels) occurs in 20–30% of hospitalized COVID-19 patients, and that cardiac injury is independently associated with adverse outcomes including mortality.
Several cardiovascular complications in COVID-19 have been identified including arrhythmia, myocardial injury, thromboembolism, and cardiomyopathy, which correlate with poorer outcomes [1, 2]. Their incidence varies significantly between study populations, with arrhythmia recognized as the second most common complication after acute respiratory distress syndrome (ARDS). In SARS-CoV‑2, the S protein is 20–30 amino acids longer, accounting for higher affinity to zinc peptidase angiotensin-converting enzyme‑2 (ACE2), found on numerous host cells including myocytes, pneumocytes, endothelial cells, and leukocytes [3, 4]. This is thought to play a crucial role in the pro-arrhythmogenic properties of SARS-CoV‑2.
Risk factors associated with increased mortality from COVID-19 include age, African American ancestry, history of preexisting cardiovascular disease [5], and comorbid conditions such as hypertension, diabetes mellitus, obesity, heart failure, renal dysfunction, and chronic lung disease [6]. Cardiac manifestations of acute COVID-19 infection include acute myocardial infarction, myocarditis leading to cardiomyopathy and potentially cardiogenic shock, bradyarrhythmias including atrioventricular block, and a plethora of supraventricular (SVT) and ventricular arrhythmias (VA; [7]). There is also growing evidence suggesting the association of COVID-19 infection with development of autonomic dysfunction leading to postural orthostatic tachycardia syndrome (POTS) and inappropriate sinus tachycardia (IST; [8]).
Mechanisms underlying arrhythmic manifestations in COVID-19 patients
Both atrial and ventricular arrhythmias can be caused by direct cardiomyocyte invasion [9] and downstream effects such as reduced expression and activity of ACE2 leading to activation of the renin–angiotensin–aldosterone axis and increased angiotensin II levels, as well as activation of cellular immune responses leading to a hyperinflammatory state and increased production of pro-inflammatory cytokines. Critically ill COVID-19 patients may develop a state called “cytokine release syndrome” (CRS), characterized clinically by rapid deterioration of respiratory status and ARDS, accompanied by signs of multiorgan dysfunction or failure, arterial and venous thromboses, and hemodynamic compromise that can progress to shock. In COVID-19 patients, CRS is characterized biochemically by elevated levels of interleukin 6 (IL-6), interleukin 8 (IL-8), interleukin 12 (IL-12), monocyte chemoattractant protein 1(MCP-1), and other biomarkers such as ferritin, fibrinogen, C‑reactive protein (CRP), D‑dimer, procalcitonin, lactate dehydrogenase (LDH), and von Willebrand factor antigen and activity [10], which makes them vulnerable to both atrial and ventricular arrhythmias.
Activation of the immune system leads to reduced regulatory T‑cell (Treg) lymphocyte number and activity, increased recruitment and activation of CD4+ CD28− T cells and differentiation into Th1 subtype of helper T cells, and increased sympathetic activation (both at the central and peripheral levels) leading to increased catecholamine levels, further increasing the potential for atrial and ventricular tachyarrhythmias. Atrial fibrillation (AF) can also occur as a result of hypoxemia, acute changes in pulmonary artery and right ventricular hemodynamics from acute pulmonary embolism or cor pulmonale, changes in atrial wall compliance and stiffness due to microvascular dysfunction and changes in atrial perfusion and contractility, and, in later stages, development of atrial fibrosis.
Patients with COVID-19-induced CRS may develop AF, which may be de novo or recurrence of preexisting paroxysmal AF due to the impact of cardiorespiratory compromise on intracardiac hemodynamics and/or electrophysiologic properties of atrial cardiomyocytes, as well as due to the development of a viral-mediated inflammatory atrial cardiomyopathy. This inflammatory cardiomyopathy is characterized by lymphocytic infiltration within the atrial cardiomyocytes, myocardial necrosis, and microangiopathic changes within the atrial vasculature.
In addition, direct viral invasion and lymphocytic infiltration of the ganglionated right atrial plexi have been reported, which may predispose to the development of AF as well as sinus node dysfunction. Finally, endothelial cell activation, as well as the activation of various elements of the clotting cascade, can lead to widespread thromboses within the pulmonary vessels (both arterial and venous), but also in atypical locations such as the right atrial appendage.
Other pathophysiological mechanisms can contribute to the development of VA and sudden cardiac arrest (SCA) in COVID-19 patients. These include ischemia due to thrombosis of small vessels within the myocardium and consequent changes in ion channel function and metabolic changes in ischemic myocytes, and QT-interval prolongation both due to increased IL‑6 levels and the use of QT-prolonging medications [11]. Other important considerations that affect the risk of VAs in COVID-19 include the following: hepatic and renal insufficiency that can cause electrolyte derangements such as hyperkalemia as well as affect metabolism and excretion of QT-prolonging medications; hyperadrenergic state related to critical illness, anxiety, or agitation due to respiratory difficulties; disruption of the sleep–wake cycle and aggravation of preexisting sleep disorders; nutritional deficiencies due to reduced appetite, prolonged ICU hospitalization, and intubated status; and the use of antimicrobials that can cause QT prolongation either individually or in combination with other antimicrobials or medications used to treat other comorbid conditions. Of note, most fatal cardiac events in COVID-19 patients are due to non-shockable rhythm disorders such as asystole or pulseless electrical activity, rather than due to VAs.
Specific arrhythmic manifestations of COVID-19
Atrial fibrillation/flutter
There is considerable evidence linking AF with states of systemic inflammation as evidenced by raised levels of inflammatory markers (CRP, IL‑6, and tumor necrosis factor‑α [TNF-α]; [12]). COVID-19 infection has been shown to cause increased levels of both CRP and IL‑6, and it is therefore not surprising that the incidence of AF is significantly higher in critically ill COVID-19 patients. In a recent retrospective study of 3970 hospitalized patients with reverse transcriptase polymerase chain reaction (RT-PCR)-confirmed COVID-19 infection, the incidence of AF/atrial flutter (AFL) was 10% (13% in a manually analyzed subset), and 4% in patients without a prior history of atrial arrhythmias. Another study of 301 critically ill patients showed that the incidence of new-onset AF was as high as 14.9% [13]. Patients with new-onset AF/AFL were older and had increased serum IL‑6 levels and greater myocardial injury (troponin‑I levels; [14]). The occurrence of AF and AFL was associated with increased mortality in this and another study [15]. A recently published retrospective analysis of hospitalized PCR-confirmed SARS-CoV‑2 infection in a single healthcare system in New York showed that AF occurred in 17.6% of these patients, of whom a majority (65.7%) had new-onset AF. In a propensity-matched analysis, AF was independently associated with a higher risk of in-hospital mortality, and new-onset AF was associated with a 56% higher risk of in-hospital mortality compared to patients with a prior history of AF [16].
The management of AF in COVID-19 patients, as in non-COVID AF patients, involves two domains: rate and/or rhythm control, and thromboprophylaxis. In patients presenting with AF or AFL with hemodynamic stability, a rate control strategy is reasonable, whereas cardioversion should be considered if hemodynamic instability occurs. Discontinuation of anti-arrhythmic drugs should be considered in patients with new-onset AF/AFL with hemodynamic stability under antiviral treatment, using rate control therapy with beta-blockers with or without digoxin. If rhythm control is desirable, then the use of amiodarone (oral or intravenous) can be considered, but close monitoring of the QTc interval on ECG, of hepatic and renal function, and of pulmonary function (as some cases of acute pulmonary toxicity with short-term use of amiodarone have been reported) is needed. Caution should also be exercised with patients on lopinavir/ritonavir, which can affect CYP3A4 function and inhibit amiodarone metabolism.
COVID-19 has been shown to induce a hypercoagulable state and can predispose patients to venous thromboembolism in the setting of acute COVID-19 infection [17]. Patients with COVID-19 and AF are thought to have a higher risk of thromboembolic complications, and young, critically ill COVID-19 patients were found to have a higher incidence of ischemic stroke [18]. Anticoagulation should be guided by the CHA2DS2-VASc score. Direct oral anticoagulants (DOACs) are preferred over vitamin K antagonists in eligible patients (i.e., those without mechanical prosthetic heart valves, moderate-to-severe mitral stenosis, or antiphospholipid syndrome) in order to avoid the need for regular determination of international normalized ratio but, considering possible drug–drug interactions, appropriate doses should be ensured [19].
There is no specific information regarding the use of DOACs in COVID-19 patients. If necessary, apixaban, edoxaban, and rivaroxaban (but not dabigatran) can be given in a crushed form. This is because dabigatran should always be given in capsule form in clinical use to avoid unintentionally increased bioavailability of dabigatran etexilate. However, if antiretroviral drugs are used, apixaban and rivaroxaban should be avoided because of potential interactions [20].
Severely ill patients may be switched to parenteral anticoagulation, given that heparin does not have significant drug–drug interactions with COVID-19 treatment (except azithromycin, which should not be co-administered with unfractionated heparin). After recovery from COVID-19, the therapeutic choices of rate or rhythm control of AF/AFL should be re-evaluated, but long-term anticoagulation should be continued based on the CHA2DS2-VASc score [19].
Atrioventricular block and sinus node dysfunction
The incidence of AV block in COVID-19 patients varies from 1% to 12% and is usually transient in nature [20]. This can manifest as high-grade second-degree AV block, such as Mobitz II second-degree AV block or 2:1 AV block, which may progress to intermittent complete heart block [21]. Many of these patients do not have preexisting cardiovascular disease or abnormalities on prior echocardiograms, and clinicians need to be aware of the association between acute COVID-19 infection and transient AV block.
Potential mechanisms for the development of AV block in COVID-19 patients include the following: focal myocarditis or myocardial edema in the region of the AV node or proximal conduction system, electrolyte disturbances, increased vagal activity during periods of endotracheal suctioning and prone positioning, and the effects of drugs including chloroquine (increases Purkinje fiber refractory period and action potential duration, resulting in AV nodal and infra-Hisian conduction disturbance), hydroxychloroquine, lopinavir/ritonavir, and azithromycin (sometimes after many weeks of treatment). Thus, patients must be informed about possible corresponding symptoms (i.e., dizziness or syncope). If persistent bradycardia occurs, all drugs that might aggravate this clinical problem should be stopped and heart rate-increasing drugs (e.g., isoprenaline or atropine) or temporary pacing must be considered. Implantation in patients with an indication for a permanent device should be delayed if possible, so as to diminish the risk of nosocomial infection.
Sinus node dysfunction leading to severe sinus bradycardia has been reported in up to 15% of cases with the SARS-CoV‑1 infection [22] and has also been reported in a few patients with COVID-19 caused by SARS-CoV‑2 [23]. The SARS-CoV‑2 virus interacts with the ACE2 receptor to enter the host cell, and the sino-atrial nodal cells are known to have a high degree of ACE2 expression [24]. The development of sinus pauses/arrest may be a reflection of the severity of the acute illness and is also a poor prognostic sign. Management of sinus node dysfunction in COVID-19 patients is similar to that for AV block as described earlier.
Ventricular arrhythmias
The incidence of ventricular arrhythmias in COVID-19 patients in large, published case series has ranged from 1.6% to 5.9% [25]. The VAs reported in COVID-19 patients include polymorphic ventricular tachycardias including torsade de pointes (TdP), ventricular tachycardia (VT) storm, and ventricular fibrillation.
Although the presence of underlying structural heart disease significantly increases the risk of VAs during acute illness, other factors that impact patients with COVID-19 may contribute to the risk of developing VA even in the absence of underlying structural heart disease. Acute myocardial injury in patients with COVID-19, as evidenced by increased cardiac troponin levels, was noted to significantly increase the risk of VA [26].
Management of VAs in patients with COVID-19 needs to employ a multipronged approach. For hemodynamically stable and unstable VA in patients with COVID-19, standard advanced cardiac life support protocols should be followed. Contributing factors such as hypoxia, hypovolemia, electrolyte abnormalities such as hypokalemia and hypomagnesemia, and metabolic acidosis should be corrected aggressively.
Vasopressor medications, sedation, and neuromuscular blockade should be employed judiciously. Other factors such as volume overload, increased sympathetic tone, and use of pro-arrhythmic drugs should be closely monitored and managed appropriately. Echocardiography to assess biventricular and valvular function should be obtained for all patients with new-onset VAs during COVID-19 infection, especially those with elevated cardiac biomarkers. Beta-blockers and/or anti-arrhythmic drug therapy should be considered for symptomatic nonsustained VA. In patients with polymorphic VT without QT prolongation, underlying myocardial ischemia should be considered as a potential cause. Intravenous (IV) beta-blockers or IV lidocaine may be used, in conjunction with intravenous sedation and use of anxiolytics in patients with respiratory distress, agitation, and anxiety. In patients who have QT prolongation and TdP, the use of QT-prolonging drugs should be stopped or modified, and IV magnesium, IV isoproterenol, or temporary transvenous pacing should be used. For patients with VT storm or recurrent or refractory VT, the use of IV amiodarone and/or lidocaine should be considered, along with deep sedation and endotracheal intubation with ventilatory support.
There are limited data supporting the use of substrate-based VT ablation in critically ill COVID-19 patients with VT storm and multiple ICD shocks [27]. Tocilizumab, a monoclonal antibody that blocks the IL‑6 receptor and is being used for critically ill COVID-19 patients, can cause QT interval shortening in association with reduction in CRP and cytokine levels and may be beneficial for use in COVID-19 patients with QT prolongation and VAs [28].
Currently, there are no data supporting the use of prophylactic anti-arrhythmic therapy for the prevention of VAs in hospitalized COVID-19 patients.
Sudden cardiac death
A worldwide survey of cardiovascular professionals early in the COVID-19 pandemic reported that cardiac arrest due to VT/VF was seen in 4.8% and pulseless electrical activity (PEA)/asystole was seen in 5.6% of hospitalized COVID-19 patients [7].
In addition, data from Italy [29] and France [30] show that the incidence of out-of-hospital cardiac arrest (OHCA) also increased significantly during the COVID-19 pandemic. The incidence of OHCA increased by up to 200% in the first few weeks of the COVID-19 pandemic in New York City compared to the same period in 2019, with higher rates of PEA (odds ratio [OR]: 1.99) and asystole (OR: 3.5) accounting for the increase in OHCA even though the prevalence of preexisting illnesses and the percentage of bystander CPR were similar between the two time periods [31].
Both bradyarrhythmias and tachyarrhythmias may be the cause of SCA in patients with COVID-19. As noted previously, critically ill COVID-19 patients with elevated cardiac troponin are at higher risk for malignant VAs and mortality. In addition, ECG abnormalities such as QRS and QTc prolongation have been observed in patients with COVID-19 pneumonia [32].
Associated with the observed increase in SCA during the COVID-19 pandemic was the alarmingly higher incidence of OHCA imposed by the shelter-in-place and lockdown restrictions. In addition, patients’ reluctance to seek medical care due to fear of contracting COVID-19 at their doctor’s office or hospital may have contributed to this. Minor causes could have been the strained emergency medical system (EMS) infrastructure due to the increased volume of activations and consequently longer EMS response times; the additional need to have access to and use personal protective equipment for first responders and front-line healthcare personnel; and the diversion of clinical personnel, administrative focus, and other resources from ongoing chronic disease management to controlling the spread of COVID-19.
Long QT syndrome in SARS-CoV-2 infection secondary to drug therapy
The antiviral properties of chloroquine by inhibition of membrane fusion were previously observed in HIV and other viruses [33]. Similarly, hydroxychloroquine was widely used with an emergency authorization [34, 35]. However, data are needed to prove their efficacy against SARS-CoV‑2 in humans. In an observational study among patients hospitalized in metropolitan New York with COVID-19, treatment with hydroxychloroquine, azithromycin, or both, compared with neither treatment, was not significantly associated with differences in in-hospital mortality [36].
In a US multicenter retrospective observational study analyzing data from 2541 patients, treatment with hydroxychloroquine alone and in combination with azithromycin was associated with a reduction in COVID-19-associated mortality when controlling for COVID-19 risk factors [35]. It should be noted that chloroquine and hydroxychloroquine prolong the QT interval and may induce life-threatening arrhythmias [37]. Thus, caution should be used in starting these agents in patients with QTc > 450 ms. Concomitant use of other QT-prolonging agents is not recommended, and abnormal electrolyte levels must be avoided. Importantly, kalemia must be maintained in a high-to-normal range.
Finally, the lack of “repolarization reserve” is of great concern, particularly if genes such as the p.Ser1103Tyr-SCN5A variant are present. In hypoxia and acidosis, there is a 10-fold increase in late sodium current activity, which in turn increases the risk of long QT syndrome, TdP, and SCD, accounting for up to 43% of deaths [38]. This puts patients with inherited channelopathies such as inherited long QT syndrome and Brugada syndrome at an increased risk of malignant arrhythmias.
Several case reports have demonstrated unmasking of Brugada syndrome by fever secondary to SARS-CoV‑2 infection [39, 40]. The Brugada ECG pattern in SARS-CoV‑2 infection and its consequences have stimulated several editorials and reviews [41, 42].
Conclusion
A variety of arrhythmic manifestations have been described in patients with COVID-19, which range from relatively benign conditions such as transient sinus bradycardia to potentially life-threatening conditions such as ventricular tachyarrhythmias and sudden cardiac death. Atrial fibrillation is the most common arrhythmia seen in acutely ill COVID-19 patients. While the pathophysiological mechanisms underlying these arrhythmias in COVID-19 patients are incompletely understood, direct viral invasion, hypoxemia, activation of systemic inflammatory systems with downstream release of cytokines and inflammatory and pro-fibrotic mediators, changes in ion channel physiology, activation of the immune system, and dysregulation of autonomic function have been implicated. Larger, multicenter epidemiological studies and randomized controlled trials are needed to truly appreciate the impact of arrhythmias, including drug-induced long QT syndrome, so as to direct further therapy in this group of patients.
References
Guzik TJ, Mohiddin SA, Dimarco A et al (2020) COVID-19 and the cardiovascular system: implications for risk assessment, diagnosis, and treatment options. Cardiovasc Res 116:1666–1687
Cheng P, Zhu H, Witteles RM et al (2020) Cardiovascular risks in patients with COVID-19: potential mechanisms and areas of uncertainty. Curr Cardiol Rep 22:34
Zhao Z, Zhang F, Xu M et al (2003) Description and clinical treatment of an early outbreak of severe acute respiratory syndrome (SARS) in Guangzhou, PR China. J Med Microbiol 52:715–720
Roden D, Harrington R, Poppas A et al (2020) Considerations for drug interactions on QTc in exploratory COVID-19 treatment. Heart Rhythm 17:231–232
Zhou F et al (2020) Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395:1054–1062
Martinez-Rubio A et al (2020) Coronavirus disease 2019 and cardiac arrhythmias. Eur Cardiol 15:e66
Gopinathannair R et al (2020) COVID-19 and cardiac arrhythmias: a global perspective on arrhythmia characteristics and management strategies. J Interv Card Electrophysiol 59:329–336
Raj SR et al (2021) Long-COVID postural tachycardia syndrome: an American Autonomic Society statement. Clin Auton Res 31:365–368
Lindner D et al (2020) Association of cardiac infection with SARS-CoV‑2 in confirmed COVID-19 autopsy cases. JAMA Cardiol 5:1281–1285
Chen T et al (2020) Clinical characteristics and outcomes of older patients with Coronavirus Disease 2019 (COVID-19) in Wuhan, China: a single-centered, retrospective study. J Gerontol A Biol Sci Med Sci 75:1788–1795
Aschauer HN et al (1988) Plasma concentrations of haloperidol and prolactin and clinical outcome in acutely psychotic patients. Pharmacopsychiatry 21:246–251
Wu N et al (2013) Association of inflammatory factors with occurrence and recurrence of atrial fibrillation: a meta-analysis. int J Cardiol 169:62–72
Ergün B et al (2021) New-onset atrial fibrillation in critically ill patients with coronavirus disease 2019 (COVID-19). J Arrhythm 37:1196–1204
Musikantow DR et al (2021) Atrial fibrillation in patients hospitalized with COVID-19: incidence, predictors, outcomes, and comparison to influenza. JACC Clin Electrophysiol 7:1120–1130
Berman JP et al (2020) Cardiac electrophysiology consultative experience at the epicenter of the COVID-19 pandemic in the United States. Indian Pacing Electrophysiol J 20:250–256
Mountantonakis SE et al (2021) Atrial fibrillation is an independent predictor for in-hospital mortality in patients admitted with SARS-CoV‑2 infection. Heart Rhythm 18:501–507
Leentjens J et al (2021) COVID-19-associated coagulopathy and antithrombotic agents—lessons after 1 year. lancet Haematol 8:e524–e533
Oxley TJ et al (2020) Large-vessel stroke as a presenting feature of COVID-19 in the young. n Engl J Med 382:e60
Martínez-Rubio A, Dan GA (2016) Cardiovascular pharmacotherapies focus: are low doses of direct-acting oral anticoagulants justified and appropriate in patients with nonvalvular atrial fibrillation? Eur Cardiol 11:115–117
Wang Y et al (2020) Cardiac arrhythmias in patients with COVID-19. J Arrhythm 36:827–836
Dagher L et al (2021) High-degree atrioventricular block in COVID-19 hospitalized patients. Europace 23:451–455
Yu CM et al (2006) Cardiovascular complications of severe acute respiratory syndrome. Postgrad Med J 82:140–144
Chinitz JS et al (2020) Bradyarrhythmias in patients with COVID-19: marker of poor prognosis? Pacing Clin Electrophysiol 43:1199–1204
Ferreira AJ et al (2011) The angiotensin-(1–7)/Mas receptor axis is expressed in sinoatrial node cells of rats. j Histochem Cytochem 59:761–768
Bhatla A et al (2020) COVID-19 and cardiac arrhythmias. Heart Rhythm 17:1439–1444
Guo T et al (2020) Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol 5:811–818
Mitacchione G et al (2020) Ventricular tachycardia storm management in a COVID-19 patient: a case report. eur Heart J Case Rep 4(FI1):1–6
Xu X et al (2020) Effective treatment of severe COVID-19 patients with tocilizumab. Proc Natl Acad Sci USA 117:10970–10975
Baldi E et al (2020) Out-of-hospital cardiac arrest during the COVID-19 outbreak in Italy. n Engl J Med 383:496–498
Marijon E et al (2020) Out-of-hospital cardiac arrest during the COVID-19 pandemic in Paris, France: a population-based, observational study. Lancet Public Health 5:e437–e443
Lai PH et al (2020) Characteristics associated with out-of hospital cardiac arrests and resuscitations during the novel coronavirus disease 2019 pandemic in New York City. JAMA Cardiol 5:1154–1163
Bertini M et al (2020) Electrocardiographic features of 431 consecutive, critically ill COVID-19 patients: an insight into the mechanisms of cardiac involvement. Europace 22:1848–1854
Savarino A, Gennero L, Chen HC et al (2001) Anti-HIV effects of chloroquine: mechanisms of inhibition and spectrum of activity. AIDS 15:2221–2229
Liu J, Cao R, Xu M et al (2020) Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV‑2 infection in vitro. Cell Discov 6:16
Mercuro NJ, Yen CF, Shim DJ et al (2020) Risk of QT interval prolongation associated with use of hydroxychloroquine with or without concomitant azithromycin among hospitalized patients testing positive for coronavirus disease 2019 (COVID-19). JAMA Cardiol 5:1036–1041
Bessière F, Roccia H, Delinière A et al (2020) Assessment of QT intervals in a case series of patients with coronavirus disease 2019 (COVID-19) infection treated with hydroxychloroquine alone or in combination with azithromycin in an intensive care unit. JAMA Cardiol 5:1067–1069
Cheung CC, Davies B, Gibbs K et al (2020) Multi-lead QT screening is necessary for QT measurement: implications for management of patients in the COVID-19 era. JACC Clin Electrophysiol 6:878–880
Giudicessi JR, Roden DM, Wilde AA, Ackerman MJ (2020) Genetic susceptibility for COVID-19-associated sudden cardiac death in African Americans. Heart Rhythm 17:1487–1492
Wu Z, McGoogan JM (2020) Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72 314 cases from the Chinese Center for Disease Control and Prevention. JAMA 323:1239–1242
Vidovich M (2020) Transient Brugada-like electrocardiographic pattern in a patient with COVID-19. J Am Coll Cardiol Case Rep 2:1245–1249
Zimmermann P, Aberer F, Braun M, Sourij H, Moser O (2022) The arrhythmogenic face of COVID-19: Brugada ECG pattern in SARS-CoV‑2 infection. J Cardiovasc Dev Dis 9:96
Sorgente A, Capulzini L, Brugada P (2020) The known into the unknown: Brugada syndrome and COVID-19. JACC Case Rep 2:1250–1251
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
K.-H. Kuck, M. Schlüter, J. Vogler, C.H. Heeger and R.R. Tilz declare that they have no competing interests.
For this article no studies with human participants or animals were performed by any of the authors. All studies mentioned were in accordance with the ethical standards indicated in each case.
Rights and permissions
About this article

Cite this article
Kuck, KH., Schlüter, M., Vogler, J. et al. Has COVID-19 changed the spectrum of arrhythmias and the incidence of sudden cardiac death?. Herz 48, 212–217 (2023). https://doi.org/10.1007/s00059-023-05186-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00059-023-05186-2