
Abstract
Pericytes play several important functions in the neurovascular unit including contractile control of capillaries, maintenance of the BBB, regulation of angiogenesis, and neuroinflammation. There exists a continuum of pericyte subtypes along the vascular tree which exhibit both morphological and transcriptomic differences. While different functions have been associated with the pericyte subtypes in vivo, numerous recent publications have used a primary human brain vascular pericytes (HBVP) cell line where this pericyte heterogeneity has not been considered. Here, we used primary HBVP cultures, high-definition imaging, cell motility tracking, and immunocytochemistry to characterise morphology, protein expression, and contractile behaviour to determine whether heterogeneity of pericytes also exists in cultures. We identified five distinct morphological subtypes that were defined using both qualitative criteria and quantitative shape analysis. The proportion of each subtype present within the culture changed as passage number increased, but pericytes did not change morphological subtype over short time periods. The rate and extent of cellular and membrane motility differed across the subtypes. Immunocytochemistry revealed differential expression of alpha-smooth muscle actin (αSMA) across subtypes. αSMA is essential for cell contractility, and consequently, only subtypes with high αSMA expression contracted in response to physiological vasoconstrictors endothelin-1 (ET1) and noradrenaline (NA). We conclude that there are distinct morphological subtypes in HBVP culture, which display different behaviours. This has significance for the use of HBVP when modelling pericyte physiology in vitro where relevance to in vivo pericyte subtypes along the vascular tree must be considered.
Graphical abstract

Similar content being viewed by others


Introduction
Pericytes are a mural cell embedded within the capillary beds of all tissues including the brain. While they were first identified over 150 years ago, many of their functions have only recently been described (Beard et al. 2020; Brown et al. 2019). Pericytes are essential for the maintenance of vascular function, particularly in the brain, where they regulate cerebral blood flow (Hall et al. 2014), maintain the blood–brain barrier (BBB) (Armulik et al. 2010; Bell et al. 2010; Daneman et al. 2010), mediate angiogenesis (Ribatti et al. 2011) and regulate immune cell trafficking and contribute to cytokine and chemokine signalling (Rustenhoven et al. 2017; Stark et al. 2013). Pericyte dysfunction and death have been implicated in the pathophysiology of many neurological diseases including stroke (Hall et al. 2014) and Alzheimer’s disease (Sagare et al. 2013; Sengillo et al. 2013), showing the importance of this cell type to brain function. However, debate in the field continues regarding the exact definition of pericytes (Attwell et al. 2016), particularly due to the significant heterogeneity in morphology and function of pericytes that has been observed at different levels of the vascular tree (Berthiaume et al. 2018b; Grant et al. 2019).
Many advancements in our understanding of pericyte functions have arisen through in vitro studies. While these studies do not incorporate the intact cerebrovascular architecture, cell culture studies provide an excellent method for interrogating the cellular physiology and the effects of stimuli and drugs directly on pericytes. Many approaches exist for isolation and culture of pericytes including primary pericytes isolated from both the rodent brain (Redzic et al. 2015; Tigges et al. 2012) and human brain (Park et al. 2022), as well as pericytes differentiated from human induced pluripotent stem cells (iPSC) (Faal et al. 2019; Gastfriend et al., 2021; Stebbins et al. 2019). Due to the limitations regarding availability, ethics and cost in sourcing primary pericytes and stem cells, as well as technical challenges required to culture them, many researchers are now utilizing commercially available sources of human pericytes, such as human brain vascular pericytes (HBVP) available from ScienCell (USA). HBVP provide a valuable pipeline for translational data given their human origin, the ability to repeat experiments easily, and culturing protocols that are well established. Our analysis of the literature for all primary human brain pericyte culture studies revealed that 64/89 (72%) of the included studies used cells sourced from ScienCell. The widespread use of HBVPs combined with the increase in popularity of pericyte research (Beard et al. 2020) highlights the importance of a comprehensive analysis of morphology and function in this primary pericyte cell source.
Recent papers have reported the use of HBVP to explore a number of pericyte functions including the PDGF-B:PDGFRβ signalling cascade (King et al. 2022; Vanlandewijck et al. 2015), pericyte-mediated vascular inflammation (Navarro et al. 2016) and pericyte contractility in response to vasoactive mediators and ischaemia (Neuhaus et al. 2017). While these functional assays are important, the heterogeneity of pericytes in vitro has recently come to light, which could impact on these functional outcomes. Park et al. (2016) showed phenotypic and functional differences in CD90+ / − subpopulations of pericytes isolated from human brain biopsy tissue, suggesting discrete roles in immune response and scar formation. In cultured pericytes derived from neurologically normal post-mortem cases, there was differential expression of several pericyte markers including alpha-smooth muscle actin (αSMA), PDGFRβ, CD146, NG2, and CD13 (Smyth et al. 2018). Therefore, we wished to determine the morphological heterogeneity of pericytes in a cell culture setting, and whether this was associated with different functions. Specifically, we investigated cell morphology and motility, gene and protein expression, and contractility in HBVP cultures that could have important implications for future studies utilising these widely used primary cells.
Methods
Cell culture
Primary human brain vascular pericytes (HBVP, #1200) were purchased from ScienCell Research Laboratories (USA), cultured in complete pericyte medium (CPM) (ScienCell, #1201) in a 37 °C incubator in standard conditions (5% CO2, 20% O2), with media changed every 2–3 days. Cells were cultured and experiments performed between passages 3 and 12, with each passage defined as the period in culture from seeding in a T75 flask with 1 million cells to ~ 90% confluence (~ 4 million cells). Passage intervals varied between 24 and 60 h. To passage, cells were washed in Dulbecco’s Phosphate Buffered Saline (DPBS) − / − (Thermofisher Scientific, #14190144), dissociated from the flask surface with TrypLE (Thermofisher Scientific, #12604013), centrifuged (200 g, 5 min), the supernatant discarded and the pellet resuspended in CPM, cells were counted, and re-plated into new T75 flasks. For experiments, HBVP were grown in T75 flasks to 90% confluence before plating onto glass coverslips (18 mm number 1 glass, Menzel-Glaser) in 12 well plates (1 mL of 5000 cells/mL in each well) for single cell imaging and immunocytochemistry (ICC), and directly into 96 well plates (100 µL of 5000 cells/mL in each well) for multicell imaging.
Imaging
For single cell imaging, high definition differential interference contrast (DIC) microscopy was performed using an upright Nikon Eclipse Ti Live Cell Microscope using a 40 × oil immersion objective with an EMCCD camera (Photometrics Evolve 512 × 512). Glass coverslips (No. 1 18 mm round, Menzel Glaser) incubated in nitric acid overnight (5 M, Merck Millipore) and UV sterilised, were washed, placed in a 12-well plate then coated with poly-L-lysine (0.01% v/v in sterile water, Sigma-Aldrich, Merck, #25988–63-0) for 1.5 h at 37 °C. Coverslips were then rinsed 3 × with CPM and plated with HBVP and allowed to settle overnight until cultures reached approximately 60–80% confluence. HBVP plated on coverslips were transferred to the microscope stage and cells were covered in imaging buffer (HBSS (no Ca2+, no Mg2+, no phenol red), 15 mM HEPES, 30 mM glucose, 1 mM MgCl2, 2 mM CaCl2 in Milli-Q H2O, pH 7.4) warmed to 37 °C. Cells were maintained at 37 °C and were protected from light. Flow of media was controlled by a gravity pump and vacuum to maintain cellular bathing and enable addition and removal of solutions while imaging. Pericytes were only imaged if there were no other cells in the field of view to allow an accurate determination of each cell’s parameters. For motility and contractility assessment, following baseline images, imaging buffer was removed and replaced with imaging buffer containing vehicle (DMSO, 0.1% v/v), noradrenaline (NA, 30 nM, Sigma #A7257) endothelin-1 (ET1, 50 nM, Sigma #E7764) or adenosine (10 µM, Sigma #A4036). Live DIC images were recorded every 1 min for 20 min. 7–12 cells per coverslip were imaged per session with an automated stage facilitating repeated return to their XY coordinates for recording over time. Images were processed with NIS Elements Analysis software and exported to ImageJ for further analysis.
To enable morphological subtype quantification over a greater number of cells, fixed cells were imaged under phase contrast at 10 × magnification from the centre of each well using a Cytation 5 Cell Imaging Multi-Mode Reader. Images were processed on Gen5 software and exported to ImageJ for further analysis.
To enable quantification of morphology over time, HBVP underwent extended live cell imaging using a temperature and humidity-controlled Nikon Eclipse Ti2-E inverted microscope with a 20 × objective. Cells were imaged every 20 min using DIC mode for 2 h. Images were processed on NIS elements software and exported as 16-bit greyscale TIFF images to ImageJ for further processing.
Immunocytochemistry
Cells were plated on coverslips and allowed to settle overnight. Cells were then fixed (ice cold 100% methanol for 10 min at − 20 °C), permeabilised and blocked with Serum Free Protein Block (Dako, Agilent, #X090930-2) containing Triton X-100 (0.3%v/v, Sigma Aldrich, #1002116296) for 1 h at room temperature. Following washing for 3 × 5 min with PBS (Gibco, #18912014), primary antibody (1:500 rabbit anti-αSMA, Abcam, ab5694) was diluted in Antibody Diluent (Dako, #S080983-2) and applied overnight in a humidified chamber at 4 °C. Cells were then washed for 3 × 5 min with PBS and incubated with 1:1000 donkey anti-rabbit AlexaFluor 488 secondary antibody (ThermoFisher, A-21206) diluted in Antibody Diluent for 1 h at room temperature. Following washing (3 × 5 min PBS then 1 × deionised water) before mounting onto slides using Prolong Gold antifade reagent with 4ʹ,6-diamidino-2-phenylindole (DAPI) mountant (Thermo Fisher, #P36931) for nuclear staining. Slides were imaged on an Olympus FV3000 Super Resolution confocal laser scanning microscope (Olympus, Japan) at 40 × magnification with detector settings as follows: DAPI (Ex. 359, Em. 461), and FITC (Ex. 498, Em. 517). Images were processed on FV-OSR software (Olympus) and exported as raw 16-bit greyscale TIFF images to ImageJ for analysis.
Pericyte morphology analysis
Each individual pericyte was manually assigned to a morphological subtype based on their appearance according to the criteria outlined in Table 1.
Images of each cell were exported to ImageJ and outlines of cells delineated using the freehand line tool. The following cell parameters were extracted: (1) cell area; (2) cell perimeter; (3) Feret’s diameter (Feret), also known as max caliper, the longest distance between any two points along the selection boundary and is normalised to perimeter, signifying cellular elongation; (4) circularity, calculated using 4π × [Area]/[Perimeter]2, with a value of 1.0 indicating a perfect circle and a value approaching 0.0 indicating an increasingly elongated cell shape. For radar plot analysis, quantified data were assigned a score of 1–5 based on their relative correlation (very low, low, moderate, high, very high) respectively to each shape descriptor in comparison to the standard pericyte. Standard pericytes made up the majority of pericytes in the cultures, and were assigned a score of 3 (moderate) for all shape descriptors.
Single cell shape analysis over time
Percentage change in cell morphology (area, perimeter, feret and circularity) was calculated after 20 min relative to baseline using the following formula, where the morphology parameter is referred to as MP:
Positive values represent an increase in the morphology measure compared to baseline and negative values represent a reduction compared to baseline. This analysis was conducted for control cells and for cells exposed to vasoactive agents NA and ET1.
Motility analysis
Cell and process motility were analysed using the MTrackJ plugin for ImageJ (Meijering et al. 2012). Cell motility was measured by tracking the movement of the cell nucleus over time, while cell process motility was analysed by tracking the movement of a primary process and a secondary process on the opposite side of the cell. For circular and sheet pericytes without primary processes, two small projections from the cell body on opposite sides to each other were selected. Velocity was calculated as µm/min. For process velocity, of the two processes, the process with the greatest velocity was used for analysis.
αSMA analysis
For analysis of αSMA expression, cell boundaries were visualised by oversaturated thresholding of the αSMA fluorescent signal. Cell boundaries were traced in ImageJ using the freehand selection tool and each cell was assigned a morphology as per criteria in Table 1. To calculate fluorescent signal per cell normalised for area and background signal, the corrected total cell fluorescence (CTCF) of cellular αSMA immunofluorescence was measured in ImageJ and calculated utilising the below formula as used previously (Jakic et al. 2017):
Statistics
All data were transferred into Microsoft Excel and then imported into GraphPad Prism (Version 9.0) for analysis. When n numbers were great enough to generate appropriate power, residuals from each data set were tested for normality using the Shapiro–Wilk normality test when n < 50, and Kolmogorov–Smirnov test when n > 50. When residuals were normally distributed, data were compared using one-way ANOVA with Dunnett’s multiple comparisons test (comparing to a single control group) or Tukey’s multiple comparisons test (comparing all groups) with a single pooled variance across multiple groups, or with an unpaired t-test with Welch’s correction to compare between two groups. When residuals were not normally distributed, data were compared using the non-parametric Kruskal–Wallis test with Dunn’s multiple comparisons test across multiple groups, or with a non-parametric Mann–Whitney or Wilcoxon matched-pairs signed rank test to compare between two groups. All data are presented as mean ± standard deviation (SD). A p < 0.05 is considered statistically significant.
Results
Cultured pericytes display five distinct morphological subtypes dependent on passage number
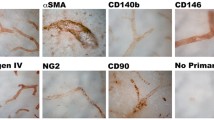
Initially, we wished to determine whether there was heterogeneity in pericyte morphology in culture. Cultured pericytes expressed the canonical pericyte markers including CD13, NG2 and PDGFRβ (Supplementary Fig. 1) as we have described before (King et al. 2022). Pericytes did not express the endothelial marker CD31 (Supplementary Fig. 1). We observed significant heterogeneity in morphology consistently across pericyte cultures (Fig. 1A), which we categorised into five morphologies: standard, circular, sheet, spindle, or balling (Table 1). Pericytes with standard morphology (Fig. 1Ai) often appeared polarised in their membrane projections, migrated readily around the culture well and are consistent with images of pericytes presented previously (Neuhaus et al. 2017). Pericytes with circular morphology (Fig. 1Aii) were less prevalent, appeared to move more slowly and smoothly, and had very active lamellipodial outer membrane movement without major membrane polarisation. Pericytes with sheet morphology (Fig. 1Aiii) appeared to be the largest in size and were much less motile, and often shared plasma membranes with each other. Pericytes with spindle morphology (Fig. 1Aiv) were elongated, had less motility, and appeared to form long-organised chains with other nearby spindle pericytes, similar to the morphology of endothelial cell cultures (Matta et al. 2007). We also observed pericytes with balling morphology (Fig. 1Av) that, following extended observation, 97% underwent cell division within 2 h of balling (Supplementary Fig. 2). In addition, we repeated these experiments in primary rat brain pericytes and a second batch of HBVP cells and showed that these same five morphologies of pericytes were also present in these cultures (Supplementary Fig. 3).

In vitro pericyte morphological heterogeneity changes with passage number. A Representative DIC images of five different morphologies observed in three different HBVP cultures: (i) standard, (ii) circular, (iii) sheet, (iv) spindle, and (v) balling morphologies. Images from cultures between P6-P8. Scale = 10 μm. B Percentage of each morphology within passage 6 cultures was calculated. N = 4 cultures and 360–400 cells analysed per culture (1528 total cells analysed). Data points are presented as individual cultures overlayed with mean ± SD. C Percentage of each morphology within cultures was calculated from passages 3–12. N = 4 cultures and 583–4625 cells analysed per passage number (20,173 total cells analysed). Residuals were normally distributed, and data for each individual subtype were compared using one-way ANOVA with Dunnett’s multiple comparisons test (comparing back to P3). Error bars are not presented on this figure for clarity
We next assessed the proportion of each pericyte morphological subtype within each culture, and whether this changes as passage number increases. Comparing broadly across passages 3 to 12, the majority of pericytes were always of standard morphology (56–73%, Fig. 1C). Within passage 6 cultures, which were the passage most commonly used in this manuscript, the majority of pericytes consisted of the standard morphology (70.87 ± 1.76%), with circular (11.77 ± 1.49%), sheet (5.70 ± 0.35%), spindle (3.54 ± 0.31%), and balling (6.00 ± 0.69%) making up the remaining cells (Fig. 1B). Similar proportions of each morphology were also observed in primary rat brain pericytes and a second batch of HBVP cells, with the majority of cells exhibiting standard morphology (Supplementary Fig. 3). Comparing across passages from the earliest passage (P3) (Fig. 1C), the proportion of standard morphology pericytes remained consistent up to P10, but was significantly decreased by P11 (P3: 69.97 ± 0.64%; P11: 57.72 ± 2.60%; p < 0.0001). Conversely, the proportion of spindle morphology pericytes began significantly increasing by P10 (P3: 5.61 ± 1.13%; P10: 10.37 ± 1.43%; p = 0.0092; Fig. 1C). There were no significant differences in proportion of circular, sheet or balling morphologies across any passages (Fig. 1C). In addition, transcriptomic analysis of pericytes at passage 9 compared to passage 5 revealed that there were 96 differentially expressed genes (Supplementary Fig. 4), which may account for changes in the percentage representation of standard and spindle morphology. Among these differentially expressed genes, gene ontology analysis suggests that genes involved in extracellular matrix organization, metabolism and biosynthesis, and symporter activity were most enriched (Supplementary Fig. 5).
Morphological subtypes of cultured pericytes possess different shape parameters
To further characterise the morphological heterogeneity of pericytes in culture, we then quantified four shape parameters for each subtype. Cells exhibiting balling morphology were excluded from this analysis as these cells nearly always went on to divide (Supplementary Fig. 2). Quantitative analysis of cell area (Fig. 2A) revealed that standard pericytes (1.00 × 104 ± 0.47 μm2) had a significantly greater cell area than circular (0.49 × 104 ± 0.14 μm2, p < 0.0001) and spindle (0.71 × 104 ± 0.30 μm2, p < 0.0001) pericytes, yet significantly smaller than sheet pericytes (1.70 × 104 ± 0.63 μm2, p < 0.0001). Circular pericytes had the smallest perimeter (Fig. 2B; 201 ± 38 μm), while sheet (513 ± 136 μm) and spindle pericytes (464 ± 133 μm) had the largest, which were all significantly different (p < 0.0001) compared to standard pericytes (386 ± 131 μm). Feret’s diameter (Feret), signifying cellular elongation (Fig. 2C), in standard pericytes (0.76 ± 0.09 μm/μm2) was significantly larger than circular (0.73 ± 0.05 μm/μm2, p = 0.0004) and sheet (0.69 ± 0.09 μm/μm2, p < 0.0001) pericytes, while spindle pericytes (0.91 ± 0.06 μm/μm2, p < 0.0001) had a significantly larger Feret’s diameter. The circularity (Fig. 2D) of standard (0.46 ± 0.17) and sheet pericytes (0.44 ± 0.16) was similar, while circular pericytes had much greater circularity (0.77 ± 0.12, p < 0.0001) and spindle pericytes had much less circularity (0.22 ± 0.10, p < 0.0001). The radar plot highlights the consolidation of different shape parameters to enable a quantifiable classification of each morphological subtype (Fig. 2E).

Morphological subtypes of cultured pericytes possess different shape parameters. Tracing around each cell provided four different shape parameters that were quantified across different pericyte morphologies. Analysis from N = 1219 cells in images of cultures between P6 and P8. (A) Cell area was the area (µm2) within the traced region of each cell. (B) Cell perimeter was calculated as the length (µm) of the trace around each cell. (C) Feret’s diameter was determined by the longest distance between any two points along the selection boundary divided by the cell perimeter, an indicator of elongation. (D) Circularity Index was defined by 4π × [Area]/[Perimeter]2 with a value of 1.0 indicating a perfect circle while a value approaching 0.0 indicating an increasingly elongated shape. Data points are presented as individual cells overlayed with mean ± SD. Kruskal–Wallis test with Dunn’s multiple comparisons test or Ordinary One-Way ANOVA with Dunnett’s multiple comparisons test was used to compare groups relative to standard morphology. *** p < 0.001, **** p < 0.0001. (E) Radar chart describing the relative correlation of each shape descriptor to each morphological subtype. Numbers (1–5) represent very low, low, moderate, high, and very high correlation, respectively
Pericyte morphology remains consistent over 2 h
Given that the proportion of pericytes with standard and spindle morphology alters between passages, we next investigated whether pericyte morphological subtypes changed over a shorter time period. Initially, we analysed pericyte morphology in live cultures over 20 min. No changes in morphology including cell area, cell perimeter, Feret’s diameter or circularity were observed for any pericyte subtype over 20 min (Fig. 3A–E). We then extended our observation time to 2 h and assessed individual pericyte morphology. During this time, pericytes were motile and migrated across the well (Fig. 3F). We did notice some small changes between subtypes of cells, e.g. a pericyte would appear to have circular morphology but cell tracking showed that the cell was initially bunched but then spread out to reveal standard morphology. Thus, it is possible that some morphological changes occur during migration. However, when we calculated the proportions of each subtype, there were no differences in overall proportions of each subtype (Fig. 3G). This suggests that pericytes do not change their morphology in a substantial way over a short time period under basal conditions.

Pericyte morphology remains consistent but pericytes are motile over 2 h. (A) Representative DIC images showing morphological subtypes at T = 0 and T = 20 min. Images from cultures between P6 and P8. Scale = 10 μm. (B–E) The change in cell area, perimeter, feret, and circularity of each pericyte subtype was calculated over 20 min. Data points are presented as individual cells overlayed with mean ± SD. Kruskal–Wallis test with Dunn’s multiple comparison test (B, C, and E) or Ordinary One-Way ANOVA with Dunnett’s multiple comparisons test (D) was used to compare groups. 96 cells were analysed in total. (F) Representative DIC images showing pericytes at T = 0 Hr (i) and T = 2 Hr (ii). Scale = 20 μm. White tracks indicate movement of individual pericytes at 20 min intervals over a 2 h period. Images from cultures between P6 and P8. (G) Percentage of four pericyte morphologies (standard, circular, sheet, and spindle) at baseline (T = 0 h) and 2 h (T = 2 h) calculated from DIC images of P6–P8 cultures. N = 256 cells analysed
Pericyte motility and process movement differs across morphological subtype
Having noted that over a 2-h period pericytes moved across the well, we next determined whether pericyte motility was associated with morphological subtype. To quantify cell movement, we tracked the nucleus (Fig. 4A), which is known to realign following cell migration (Friedl et al. 2011), for each pericyte. Analysis of cell velocity (Fig. 4B) revealed that standard pericytes were significantly more motile (2.01 ± 0.79 μm/min) compared to both sheet (1.12 ± 0.32 μm/min, p < 0.0001) and spindle (0.79 ± 0.29 μm/min, p < 0.0001) pericytes, but were not significantly more motile than circular pericytes (1.45 ± 0.54 μm/min, p = 0.1007). In addition, we quantified process velocity through tracking the most active of two points on the membrane at the sites of primary projections (Fig. 4C). Process velocity was significantly greater in standard pericytes (2.7 ± 1.31 μm/min) compared to both sheet (1.41 ± 0.64 μm/min, p < 0.0001) and spindle (1.42 ± 0.97 μm/min, p < 0.0001) pericytes, while there was no difference with circular pericytes (2.30 ± 0.77 μm/min). Together, these data suggest that standard and circular pericytes are motile subtypes, whereas sheet and spindle pericytes have reduced cellular and process motility. As process motility is an important function of both contraction and relaxation of pericytes to modulate blood vessel tone, there may be differences between pericyte subtypes in the responsiveness to vasoactive cues.

Pericyte cell velocity and process velocity differs across morphological subtype. (A) Representative DIC images showing morphological subtypes at T = 0 and T = 20 min. Tracks indicate 1 min interval movement of the cell nucleus (pink, 1) and most active membrane projection (cyan, 2). Images from cultures between P6 and P8. (B) Mean cell velocity as calculated by the distance of the pink tracks over time in different pericyte morphological subtypes. (C) Process velocity in different pericyte morphological subtypes was assessed in the most active process determined by the highest velocity process per cell out of the two primary processes measured as indicated by the cyan tracks. 106 cells were analysed in total. Data points are presented as individual cells overlayed with mean ± SD. Kruskal–Wallis test with Dunn’s multiple comparisons test was used to compare groups. **** p < 0.0001, *** p < 0.001 compared to Standard
Pericyte contractility differs across morphological subtype
Pericytes have previously been shown to be contractile in vitro (Neuhaus et al. 2017) and their contractility in vivo appears to be restricted to the ensheathing pericytes at the arteriolar end of the capillary bed (Gonzales et al. 2020; Hartmann et al. 2022). Here, after determining morphological subtype differences in pericyte motility, we investigated whether contractility also differed between subtypes. First, we assessed whether pericytes expressed the receptors for the vasoconstrictors NA and ET1. Expression of the NA receptor α1B-Adrenergic receptor (α1B) was highly expressed in pericytes, while expression of the endothelin A receptor (ETA) revealed a more punctate staining pattern (Supplementary Fig. 6A). Following 20-min exposure to the vasoconstrictor NA, standard pericytes responded most strongly through process retraction and a reduction in cell area, while minimal changes occurred in other subtypes (Fig. 5A). Quantification (Fig. 5B) showed that NA significantly reduced cell area in standard pericytes from baseline (− 47.4 ± 20.1%) compared to control treatment (− 3.7 ± 25.7%, p < 0.0001; Fig. 5B). There were no significant changes in cell area in circular, sheet or spindle pericytes compared to controls. Similar results were obtained with the vasoconstrictor ET1 (Fig. 5C), whereby standard pericytes had reduced cell area from baseline with ET1 (− 22.6 ± 30.5%) compared to control conditions (− 3.7 ± 25.7%, p = 0.0016), with no significant change in other subtypes (Fig. 5C). The extent of pericyte contraction was greater with NA compared to ET1. To confirm that cell contraction was not a result of the initiation of cell death, we performed a cell death assay, which showed that exposure to NA and ET1 did not lead to a significant increase in cell death over 1 h (Supplementary Fig. 6B). To determine whether standard pericytes could also relax, adenosine, a known vasorelaxant, was administered following pericyte contraction with NA. Exposure of pericytes previously contracted with NA to adenosine revealed a restoration above baseline cell area, indicating pericytes could also relax (Supplementary Fig. 7). These data suggest that standard pericytes are the most responsive to vasoactive cues.

Standard morphology pericytes contract in response to NA and ET1. (A) Representative panel of DIC images of standard (i), circular (ii), sheet (iii), and spindle (iv) pericytes at T = 0 and T = 20 min treated with NA. The white dotted outline indicates tracing of the cell area. Images from cultures between P6 and P8. (B–C) Relative change in cell area compared to baseline area following exposure to NA (B) and ET1 (C). Control indicates treatment with a DMSO vehicle. 233 cells were analysed in total. Data points are presented as individual cells overlayed with mean ± SD. Kruskal–Wallis test with Dunn’s multiple comparisons test was used to compare groups. **** p < 0.0001, *** p < 0.001, ** p < 0.001, * p < 0.05 compared to Standard in either control or treatment group
Pericyte αSMA expression differs across morphological subtype
One of the key proteins involved in cytoskeletal rearrangement during cell contraction is αSMA (Hinz et al. 2001; Wang et al. 2006). Given that we observed differences in the extent of contractility between pericyte subtypes, we hypothesised that this may be due to differing expression of the key cell contractility protein αSMA. Immunostaining showed HBVP cultures had widespread expression of αSMA protein and we found variations in αSMA staining intensity between cells and between subtypes (Fig. 6A). We used CTCF to quantify fluorescence intensity of αSMA to determine any differences in between subtypes. Standard pericytes had the greatest level of αSMA expression (7.64 × 105 ± 6.03 × 105 AU) which was significantly higher than both circular (3.68 × 105 ± 9.32 × 104 AU, p < 0.0001) and spindle (4.23 × 105 ± 1.28 × 105 AU, p < 0.0001) pericytes. Standard pericytes also had higher αSMA expression than sheet pericytes (7.42 × 105 ± 2.49 × 105 AU, p = 0.0170), though levels were still moderately high in sheet pericytes compared to circular and spindle (Fig. 6B). Interestingly, sheet pericytes also had some level of contraction compared to baseline following NA administration (− 16.00 ± 11.39%; Fig. 5B). These data suggest that the differential contractile capabilities of pericyte subtypes may be associated with each subtype’s αSMA expression.

αSMA expression in pericytes varies across morphological subtype. (A) Representative confocal microscopy images of pericyte subtypes with αSMA-positive (green) and DAPI-positive nuclei (blue). White dotted outlines represent cell membrane boundaries. Images from P6 cultures. (B) Quantification of corrected total cell fluorescence (CTCF) of αSMA expression between pericyte subtypes. 1219 cells were analysed in total. Data points are presented as individual cells overlayed with mean ± SD. Kruskal–Wallis test with Dunn’s multiple comparisons test was used to compare groups. **** p < 0.0001, * p < 0.05 compared to standard
Discussion
After close examination of HBVP by DIC microscopy, here we report five major subtypes of pericyte morphology in culture. The majority of pericytes in culture (around 70%) exhibited ‘standard’ morphology and the remaining four morphologies were in similar proportions to each other (5–12% of all cells). Cell tracing analysis identified key shape descriptors that could describe each morphology, and cell tracing technology could be used to distinguish morphological subtype of pericytes to ensure consistency of cultures throughout all experiments. While heterogeneity of pericytes on blood vessels in vivo is thought to be dependent on location of the vascular tree (Berthiaume et al. 2018b; Grant et al. 2019), pericytes cultured in a monolayer in vitro do not have tubular structures to adhere to or in vivo cues to interact with such as factors released from endothelial cells (Smyth et al. 2018), suggesting that other factors may produce the morphological and functional differences of pericytes within a culture. The development of 3D co-cultures mimicking a tubular blood vessel structure could lead to similarities in pericyte morphologies between in vitro and in vivo studies (Campisi et al. 2018; Kim et al. 2015). Here we highlight that differential pericyte morphology in cultured HBVPs is associated with differences in motility, contractility and protein expression. Importantly, standard pericytes were the most contractile subtype with the greatest expression of αSMA, highlighting the importance of protein expression to function within individual cells of pericyte cultures. Therefore, the implications of cellular heterogeneity within cultures need to be considered when assessing pericyte function.
Passage number is critical for the use of pericytes in culture as phenotype and gene expression may change over time, as has been shown previously with placental pericytes (Shiwen et al. 2009). Between passages 3 and 9, the proportions of each subtype remained similar, suggesting that morphological heterogeneity displayed within cultures was not related to cell passage. Passage 9 represents approximately 18 population doublings which supports the recommendation from ScienCell for the use of HBVPs up to 15 population doublings. However, from passage 10 the proportion of standard pericytes significantly reduced while the proportion of spindle cells increased. Spindle pericytes appeared to be less motile and did not proliferate as readily as other morphologies. Interestingly, pericytes in the aged mouse brain have reduced function including diminished remodelling capacity and dynamic control over blood vessels in vivo (Berthiaume et al. 2022). Cell morphology changes have been observed with greater passage number for other cell types (Neumann et al. 2010; Yang et al. 2018) restricting experiments to specific passage number containing optimal cells. Our RNAseq data highlighted a number of differentially expressed genes between passages 5 and 9, many of which were involved in extracellular matrix organization, metabolism and biosynthesis, and symporter activity. This indicates that to maintain consistent cultures, experiments should be performed in HBVP within defined passage numbers at similar cell densities.
Both media and confluency of cells may have influenced morphology and function of cultured pericytes. Our studies on HBVP were conducted using complete pericyte media sourced from ScienCell, whose formulation is proprietary. A previous study has assessed the phenotype and neuroinflammatory responses of primary human pericytes exposed to pericyte media from ScienCell compared to DMEM/F12 media (Rustenhoven et al. 2018). This paper noted differences in pericyte proliferation, morphology, protein expression, and phagocytic and migratory ability between both media types. These phenotypic changes were assessed between cultures, and do not account for any pericyte heterogeneity within cultures as observed in our study. Our rat brain pericyte analysis showed that similar morphological heterogeneity exists in cultures that were grown in DMEM-based media, and it will be important for future studies to ensure that morphological and functional heterogeneity exists in different media formulations within a culture. In addition, our experiments were conducted at approximately 60–80% confluency to enable individual pericyte visualization using single cell imaging. While most studies appear to use a confluency > 80%, a very high confluency can lead to contact inhibition which can arrest cell growth (Seluanov et al. 2009) and initiate cell death due to depletion of nutrients (Busschots et al. 2015). Therefore, it is possible that the heterogeneity in both morphology and function of cultured pericytes may change in a high confluency environment.
While the proportion of morphological subtypes changed with later passage, imaging over a 2-h period showed that morphology of individual cells did not change quickly under basal conditions. Single pericyte ablation studies have shown that it takes many days for pericytes to morphologically adapt to cover endothelial cells on the capillary, suggesting morphological change of in vivo pericytes is a slow process (Berthiaume et al. 2018a). Tracking cell and process velocity over time showed that in culture, standard pericytes were the most active, with extension and contraction of their processes. This aligns with capillary pericytes in vivo where extension and retraction of processes have been shown to aid in the coverage of the vasculature and sensing of molecules from within the circulation and the brain parenchyma (Berthiaume et al. 2022). It is possible that differences in morphology could represent different states of the cell cycle. It is known that as mammalian cells enter different stages of the cell cycle, their morphology changes (Thery and Bornens 2006). Our morphology analysis highlighted balled cells as cells about to undergo cell division, and we identified that standard, circular and sheet cells all underwent cell division. Further research is needed to confirm whether morphological subtypes of pericytes represent different stages of the cell cycle.
One of the key functions of pericytes in the brain is to regulate cerebral blood flow (Hall et al. 2014), but it is thought that only ensheathing pericytes on the arteriolar end of the capillary bed are able to contract and relax capillaries (Attwell et al. 2016). Previous in vitro and ex vivo studies have shown that pericytes can contract or relax in response to vasoactive mediators (Hall et al. 2014; Neuhaus et al. 2017), but knowledge on whether the pericyte response to these mediators varies with morphological subtype is limited. Here, we showed that standard pericytes contracted most strongly in response to both NA and ET1 compared to other subtypes. Given that standard pericytes make up the majority subtype in HBVP cultures, this may explain why contraction and relaxation still occurred in whole-well assays (Neuhaus et al. 2017). However, some sub-population differences within cultured pericytes for Ca2+ release (Halaidych et al. 2019) and cell contraction (Heyba et al. 2019) were observed following ET1 stimulation. We then explored whether there were differences in αSMA expression between morphological subtypes. αSMA is a critical protein mediating the contraction of vascular smooth muscle cells (Wang et al. 2006), and has been shown to be expressed at differing levels in pericytes in vivo (Attwell et al. 2016). Our subtype analysis revealed that pericytes with standard morphology expressed αSMA most strongly, which was associated with the most profound contraction. Other morphological subtypes expressed αSMA to a lower level but did not exhibit contraction in response to NA or ET1. Future studies should look at vasoactive mediator receptor expression in the different pericyte subtypes as differential NA or ET1 receptor expression levels may lead to the differences in contractility we observed. One limitation of using cell retraction, a method to measure pericyte contractility which has been used previously (Kelley et al. 1987; Neuhaus et al. 2017), is cell retraction or loss of cell area could be due to other factors such as apoptosis or trypsinization. We have attempted to overcome this by showing no cell death occurred with NA or ET1 administration, and pericytes could actively relax (increase their cell area) in response to adenosine. Other contraction assays would be needed to confirm these effects.
There were a few other limitations to our study. Our initial classification of morphological subtypes of pericytes was performed manually, but the shape descriptors following cell tracing revealed clear differences between subtypes. We also only imaged cells up to 2 h, and longer imaging times could determine if changes in morphology occur more frequently. Additional studies may also use FACS to distinguish phenotypic heterogeneity in pericytes similar to that previously used for cell cycle analysis in HEK293 cells (Kage et al. 2021). In addition, single cell transcriptomics could be applied on FACS sorted cells to uncover molecular characteristics of each morphological subtype.
With the current evidence, it is difficult to conclude which in vitro subtype of pericyte corresponds to the subtype of pericyte in vivo. Brain pericytes on capillary beds have various morphology, protein and mRNA expression, and functions depending on their location in the vascular tree. For example, expression of αSMA is thought to be associated with contractile properties in ensheathing pericytes (Attwell et al. 2016), with limited expression in other pericyte subtypes (Vanlandewijck et al. 2018). However, Smyth et al. (2018) showed some αSMA expression on junctional pericytes in vivo, while Alarcon-Martinez et al. (2018) showed that capillary pericytes do express αSMA which rapidly depolymerizes during tissue fixation thus evading detection by immunolabeling. While we showed that HBVPs had heterogeneous αSMA expression, expression of αSMA in in vitro pericytes has been shown previously, which may be due to upregulation of αSMA expression in culture conditions (Heyba et al. 2019; Smyth et al. 2018). Here, we only assessed whether pericyte subtypes had differing motility and contractility, and further analysis of other pericyte functions such as BBB integrity, immune activity and susceptibility to cell death would need to be carried to determine the full scope of functional heterogeneity between morphological subtypes of pericytes, and whether there was an association with an in vivo subtype.
In conclusion, we have identified distinct morphological subtypes in pericyte cultures, which display different behaviours. The predominant standard morphology had the greatest cell and process motility, and contractile response to vasoactive mediators. Morphological diversity within cell culture systems is important to consider when performing assays, considering that the proportion of morphological subtypes in pericyte cultures change as passage number increases, which may lead to different functional properties. Given that pericytes in the brain have substantial morphological and functional differences depending on where they sit within the vascular tree, it is important to consider and understand pericyte heterogeneity in vitro in order to inform brain pericyte biology.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Alarcon-Martinez L et al. Capillary pericytes express alpha-smooth muscle actin, which requires prevention of filamentous-actin depolymerization for detection. Elife. 2018;7. https://doi.org/10.7554/eLife.34861
Armulik A, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–61. https://doi.org/10.1038/nature09522.
Attwell D, et al. What is a pericyte? J Cereb Blood Flow Metab. 2016;36:451–5. https://doi.org/10.1177/0271678X15610340.
Beard DJ, et al. The rise of pericytes in neurovascular research. J Cereb Blood Flow Metab. 2020;40:2366–73. https://doi.org/10.1177/0271678X20958497.
Bell RD, et al. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68:409–27. https://doi.org/10.1016/j.neuron.2010.09.043.
Berthiaume AA, et al. Dynamic remodeling of pericytes in vivo maintains capillary coverage in the adult mouse brain. Cell Rep. 2018a;22:8–16. https://doi.org/10.1016/j.celrep.2017.12.016.
Berthiaume AA, et al. Pericyte structural remodeling in cerebrovascular health and homeostasis. Front Aging Neurosci. 2018b;10:210. https://doi.org/10.3389/fnagi.2018.00210.
Berthiaume AA, et al. Pericyte remodeling is deficient in the aged brain and contributes to impaired capillary flow and structure. Nat Commun. 2022;13:5912. https://doi.org/10.1038/s41467-022-33464-w.
Brown LS, et al. Pericytes and neurovascular function in the healthy and diseased brain. Front Cell Neurosci. 2019;13:282. https://doi.org/10.3389/fncel.2019.00282.
Busschots S, et al. Non-invasive and non-destructive measurements of confluence in cultured adherent cell lines. MethodsX. 2015;2:8–13. https://doi.org/10.1016/j.mex.2014.11.002.
Campisi M, et al. 3D self-organized microvascular model of the human blood-brain barrier with endothelial cells, pericytes and astrocytes. Biomaterials. 2018;180:117–29. https://doi.org/10.1016/j.biomaterials.2018.07.014.
Daneman R, et al. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468:562–6. https://doi.org/10.1038/nature09513.
Faal T, et al. Induction of mesoderm and neural crest-derived pericytes from human pluripotent stem cells to study blood-brain barrier interactions. Stem Cell Reports. 2019;12:451–60. https://doi.org/10.1016/j.stemcr.2019.01.005.
Friedl P, et al. Nuclear mechanics during cell migration. Curr Opin Cell Biol. 2011;23:55–64. https://doi.org/10.1016/j.ceb.2010.10.015.
Gastfriend BD, et al. Differentiation of brain pericyte-like cells from human pluripotent stem cell-derived neural crest. Curr Protoc. 2021;1:e21. https://doi.org/10.1002/cpz1.21
Gonzales AL, et al. Contractile pericytes determine the direction of blood flow at capillary junctions. Proc Natl Acad Sci U S A. 2020;117:27022–33. https://doi.org/10.1073/pnas.1922755117.
Grant RI, et al. Organizational hierarchy and structural diversity of microvascular pericytes in adult mouse cortex. J Cereb Blood Flow Metab. 2019;39:411–25. https://doi.org/10.1177/0271678X17732229.
Halaidych OV, et al. Quantifying Ca(2+) signaling and contraction in vascular pericytes and smooth muscle cells. Biochem Biophys Res Commun. 2019;513:112–8. https://doi.org/10.1016/j.bbrc.2019.03.143.
Hall CN, et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 2014;508:55–60. https://doi.org/10.1038/nature13165.
Hartmann DA, et al. Pericyte control of blood flow across microvascular zones in the central nervous system. Annu Rev Physiol. 2022;84:331–54. https://doi.org/10.1146/annurev-physiol-061121-040127.
Heyba M, et al. Viability and contractility of rat brain pericytes in conditions that mimic stroke; an in vitro study. Front Neurosci. 2019;13:1306. https://doi.org/10.3389/fnins.2019.01306.
Hinz B, et al. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol Biol Cell. 2001;12:2730–41. https://doi.org/10.1091/mbc.12.9.2730.
Jakic B, et al. Elevated sodium leads to the increased expression of HSP60 and induces apoptosis in HUVECs. PLoS One. 2017;12:e0179383. https://doi.org/10.1371/journal.pone.0179383.
Kage D, et al. Multi-angle pulse shape detection of scattered light in flow cytometry for label-free cell cycle classification. Commun Biol. 2021;4:1144. https://doi.org/10.1038/s42003-021-02664-3.
Kelley C, et al. Microvascular pericyte contractility in vitro: comparison with other cells of the vascular wall. J Cell Biol. 1987;104:483–90. https://doi.org/10.1083/jcb.104.3.483.
Kim J, et al. Engineering of a biomimetic pericyte-covered 3D microvascular network. PLoS One. 2015;10:e0133880. https://doi.org/10.1371/journal.pone.0133880.
King NE, et al. Pharmacological PDGFRbeta inhibitors imatinib and sunitinib cause human brain pericyte death in vitro. Toxicol Appl Pharmacol. 2022;444:116025. https://doi.org/10.1016/j.taap.2022.116025.
Matta H, Surabhi R M, Zhao J, Punj V, Sun Q, Schamus S, Mazzacurati L, Chaudhary P M Induction of spindle cell morphology in human vascular endothelial cells by human herpesvirus 8-encoded viral FLICE inhibitory protein K13 Oncogene 2007;26(11):1656-1660. https://doi.org/10.1038/sj.onc.1209931
Meijering E, et al. Methods for cell and particle tracking. Methods Enzymol. 2012;504:183–200. https://doi.org/10.1016/B978-0-12-391857-4.00009-4.
Navarro R, et al. Immune regulation by pericytes: modulating innate and adaptive immunity. Front Immunol. 2016;7:480. https://doi.org/10.3389/fimmu.2016.00480.
Neuhaus AA, et al. Novel method to study pericyte contractility and responses to ischaemia in vitro using electrical impedance. J Cereb Blood Flow Metab. 2017;37:2013–24. https://doi.org/10.1177/0271678X16659495.
Neumann E, et al. Cell culture and passaging alters gene expression pattern and proliferation rate in rheumatoid arthritis synovial fibroblasts. Arthritis Res Ther. 2010;12:R83. https://doi.org/10.1186/ar3010.
Park TI, et al. Cultured pericytes from human brain show phenotypic and functional differences associated with differential CD90 expression. Sci Rep. 2016;6:26587. https://doi.org/10.1038/srep26587.
Park TI, et al. Routine culture and study of adult human brain cells from neurosurgical specimens. Nat Protoc. 2022;17:190–221. https://doi.org/10.1038/s41596-021-00637-8.
Redzic ZB, et al. Differential effects of paracrine factors on the survival of cells of the neurovascular unit during oxygen glucose deprivation. Int J Stroke. 2015;10:407–14. https://doi.org/10.1111/ijs.12197.
Ribatti D, et al. The role of pericytes in angiogenesis. Int J Dev Biol. 2011;55:261–8. https://doi.org/10.1387/ijdb.103167dr.
Rustenhoven J, et al. Brain pericytes as mediators of neuroinflammation. Trends Pharmacol Sci. 2017;38:291–304. https://doi.org/10.1016/j.tips.2016.12.001.
Rustenhoven J, et al. Modelling physiological and pathological conditions to study pericyte biology in brain function and dysfunction. BMC Neurosci. 2018;19:6. https://doi.org/10.1186/s12868-018-0405-4.
Sagare AP, et al. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat Commun. 2013;4:2932. https://doi.org/10.1038/ncomms3932.
Seluanov A, et al. Hypersensitivity to contact inhibition provides a clue to cancer resistance of naked mole-rat. Proc Natl Acad Sci U S A. 2009;106:19352–7. https://doi.org/10.1073/pnas.0905252106.
Sengillo JD, et al. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer’s disease. Brain Pathol. 2013;23:303–10. https://doi.org/10.1111/bpa.12004.
Shiwen X, et al. Pericytes display increased CCN2 expression upon culturing. J Cell Commun Signal. 2009;3:61–4. https://doi.org/10.1007/s12079-009-0053-7.
Smyth LCD, et al. Markers for human brain pericytes and smooth muscle cells. J Chem Neuroanat. 2018;92:48–60. https://doi.org/10.1016/j.jchemneu.2018.06.001.
Stark K, et al. Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and “instruct” them with pattern-recognition and motility programs. Nat Immunol. 2013;14:41–51. https://doi.org/10.1038/ni.2477.
Stebbins MJ, et al. Human pluripotent stem cell-derived brain pericyte-like cells induce blood-brain barrier properties. Sci Adv. 2019;5:eaau7375. https://doi.org/10.1126/sciadv.aau7375.
Thery M, Bornens M. Cell shape and cell division. Curr Opin Cell Biol. 2006;18:648–57. https://doi.org/10.1016/j.ceb.2006.10.001.
Tigges U, et al. A novel and simple method for culturing pericytes from mouse brain. Microvasc Res. 2012;84:74–80. https://doi.org/10.1016/j.mvr.2012.03.008.
Vanlandewijck M, et al. Functional characterization of germline mutations in PDGFB and PDGFRB in primary familial brain calcification. PLoS One. 2015;10:e0143407. https://doi.org/10.1371/journal.pone.0143407.
Vanlandewijck M, et al. Author correction: a molecular atlas of cell types and zonation in the brain vasculature. Nature. 2018;560:E3. https://doi.org/10.1038/s41586-018-0232-x.
Wang J, et al. Multiple roles of alpha-smooth muscle actin in mechanotransduction. Exp Cell Res. 2006;312:205–14. https://doi.org/10.1016/j.yexcr.2005.11.004.
Yang YK, et al. Changes in phenotype and differentiation potential of human mesenchymal stem cells aging in vitro. Stem Cell Res Ther. 2018;9:131. https://doi.org/10.1186/s13287-018-0876-3.
Acknowledgements
The authors wish to acknowledge the use of the services and facilities of the Australian Genome Research Facility for RNA sequencing and analysis. We would also like to acknowledge Dr Tamer Rabie (University of Central Lancashire, UK) for providing the rat brain pericyte images for analysis.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions This research was supported by funding from the National Health and Medical Research Council of Australia (APP1137776 (BAS), APP1163384 (BAS), APP2003351 (BAS, DWH)), Rebecca Cooper Foundation (BAS) and the University of Tasmania (DWH, LSB).
Author information
Authors and Affiliations
Contributions
LSB: Conceptualization, Methodology, Software, Validation, Formal Analysis, Investigation, Data Curation, Writing—Original Draft, Visualization; NEK: Methodology, Validation, Investigation, Writing—Review & Editing; JMC: Methodology, Software, Investigation, Resources, Data Curation, Writing—Review & Editing; RJG: Conceptualization, Methodology, Validation, Resources, Writing—Review & Editing; LF: Conceptualization, Resources, Writing—Review & Editing, Supervision; DWH: Conceptualization, Writing—Review & Editing, Supervision, Funding Acquisition; BAS: Conceptualization, Writing—Original Draft, Visualization, Supervision, Project Administration, Funding Acquisition.
Corresponding author
Ethics declarations
Ethical approval
Acquiring and performing cell culture experiments on primary human cells from a commercial source conforms to the National Statement on Ethical Conduct in Human Research, National Health and Medical Research Council (NHMRC) of Australia, and does not require specific institutional ethics approval.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brown, L.S., King, N.E., Courtney, JM. et al. Brain pericytes in culture display diverse morphological and functional phenotypes. Cell Biol Toxicol 39, 2999–3014 (2023). https://doi.org/10.1007/s10565-023-09814-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10565-023-09814-9