
Abstract
In the first two papers of this series [Behrens, Phys Chem Minerals 48:8, 2021a; Behrens, Phys Chem Minerals 48:27, 2021b], incorporation of hydrogen in the feldspar structure, partitioning of hydrogen between feldspars and gases/fluids and self-diffusion of hydrogen in feldspars have been discussed, with particular focus on sanidine. Here, the results of reactions between sanidine containing strongly bonded hydrogen defects and (Na,K)Cl are presented. Experiments were performed at ambient pressure at temperatures of 605–1000 °C, and hydrogen profiles were measured by IR microspectroscopy. Profiles can be interpreted by an incomplete dehydrogenation at the crystal surface or a strong concentration dependence of hydrogen diffusivity. Both are consistent with hydrogen located on interstitial sites and difficult to substitute by the larger alkali ions. Chemical diffusivities of hydrogen derived from fitting of the profiles or Boltzmann–Matano analysis are similar to self-diffusivities determined by D/H exchange experiments. Activation energies are also comparable. Comparison to sodium and potassium diffusion data for sanidine (Wilangowski et al. in Defect Diffus Forum 363: 79–84, 2015; Hergemöller et al. in Phys Chem Minerals 44:345–351, 2017) supports a mechanism of proton diffusion charge-compensated by Na+ diffusion for hydrogen removal in the sanidines under dry conditions.
Similar content being viewed by others

Introduction
Hydrogen is a common impurity in nominally anhydrous minerals, such as olivine, pyroxene, feldspar or quartz (e.g. Beran and LiIbowitzky 2006; Johnson 2006; Skogby 2006). The incorporated hydrogen can have major impacts on the properties of the minerals. In the case of feldspars, diffusion rates of network-forming elements, such as silicon, aluminium and oxygen, are strongly enhanced by dissolved hydrogen species (e.g. Yund and Anderson 1974; Giletti et al. 1978; Grove et al. 1984; Yund 1986; Elphick et al. 1988; Farver and Yund 1990; Baschek und Johannes 1995; Cherniak 2010; Farver 2010). The enhancement of intracrystalline diffusion is important not only on the local scale, but it also affects long-range processes in rocks, i.e. the tectonic response to accumulating stress induced by plate movement or the mobilization of elements in rocks as a prerequisite for the formation of ore deposits. Thus, there is large interest in better understanding the properties of dissolved hydrogen in feldspars.
In feldspars formed or recrystallized under metamorphic conditions, most of the hydrogen is typically present in inclusions. These can be fluid inclusions as well as hydrous glasses or minerals. Regarding the kinetic properties of feldspars, such localized hydrogen pools have little impact, while hydrogen species incorporation as point defects in the feldspar structure are more important. Using polarized IR spectroscopy, Johnson and Rossman (2003, 2004) identified different types of structural hydrogen defects in feldspar. According to their studies, volcanic feldspars contain only structural OH groups while plutonic and pegmatitic feldspars may also host other hydrogen species, i.e. H2O molecules and ammonium cations. A common OH defect in alkali feldspars and plagioclase, denoted as type IIa OH bands by Johnson and Rossman (2004), is characterized by a broad IR band system centred around 3300 cm−1 with the maximum intensity closely aligned with the crystallographic a axis.
A different IR absorption spectrum is observed for Eifel sanidine with a band at 3050 cm−1 is intense in the b direction, while a band near 3400 cm−1 is most intense in the a direction. Johnson and Rossman (2004) classified the corresponding defect in Eifel sanidines as type IIb OH. However, results of the experimental studies of Behrens (2021a, b) suggest that hydrogen incorporation in Eifel sanidines is more complex and at least two different OH defects with different mobilities coexist in the structure.
This paper is the third part of a series of studies examing hydrogen defects in feldspar. In the first part, the thermal stability of hydrogen defects in feldspars and the partitioning of water between a gas phase and feldspars (plagioclase and K-rich alkali feldspar) was investigated at ambient and elevated pressure (Behrens 2021a). Diffusion of hydrogen species and hydrogen isotope exchange between coexisting fluid/gas phases was the main topic of the second part (Behrens 2021b). Different transport mechanisms for hydrogen are indicated at wet and at dry conditions. At elevated water pressures, water molecules act as transport vehicles for hydrogen. Under dry conditions, diffusion of protons resulting from dissociation of extrinsic defects such as AlOH groups is the dominant process.
The latter mechanism is the focus of the third paper of this series. Dehydrogenation experiments were performed with sanidine in contact with alkali chloride. Behrens (2021a) demonstrated that the release of hydrogen from natural sanidine is strongly accelerated when alkali ions are available for charge compensation of out-diffusing protons. Here, this process is investigated in detail over a large temperature range. The data are compared to self- diffusion of hydrogen determined by isotope exchange experiments (Behrens 2021a) and alkali diffusion data (Neusser et al. 2012; Schäffer et al. 2014; Wilangowski et al. 2015; Hergemöller et al. 2017) to constrain the mechanisms of dehydrogenation.
Samples and experiments
Cuboids of sanidine were used to probe hydrogen diffusion in three orthogonal directions. The sanidine SV has gemstone quality and is described in detail in the first paper (Behrens 2021a). Based on cleavage planes in (010) and (001), crystals were oriented. The pleochroisms of the OH vibration bands at 3400 cm−1 and 3050 cm−1 in polarized IR absorption spectra give further constraints of the crystallographic axis in the cuboid. The crystals were heated for at least 7 days in air at 900 °C to remove large portions of the mobile type OH IIa defects.
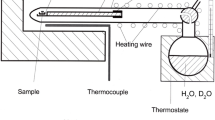
Experiments were performed at ambient pressure in a large temperature range from 605 to 1000 °C (Table 1). At temperatures below 800 °C, the samples were embedded in solid alkali chloride. Some of these experiments were performed in a closed glass apparatus, similar to that used for D-H exchange experiments (Behrens 2021b, Fig. 1). A dry atmosphere was set by evacuating and filling the apparatus several times with air pre-dried over phosphorus pentoxide. In other experiments, the salt–sanidine assembly was loaded in a Pt-boat and continuously flushed with a pre-dried air stream.

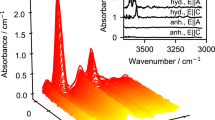
a, b Examples of hydrogen profiles in sanidine after desorption experiments with alkali chloride. Lines represent fits by Eq. (1), i.e. assuming constant diffusion coefficients
At temperatures above the melting points of the alkali chlorides, closed gold capsules were used. Either the cuboid was placed directly in the melt, or it was set on a small table above the melt so that the sample is only in contact with the vapour phase. This set-up was shown in Fig. 9 of Behrens (2021b).
Alkali chlorides consist of pure NaCl, pure KCl or K0.95Na0.05Cl. NaCl was chosen because of the high mobility of Na+ in sanidine (Wilangowski et al. 2015) and because the smaller size of Na+ compared to K+ favours substitution for H+ on interstitial sites (Behrens 2021a). However, a serious disadvantage of using NaCl is that the cation exchange in the sanidine causes severe stress and cracking (Scheidl et al. 2013; Petrishcheva et al. 2019). Therefore, K-rich chlorides were used in most of the experiments. At the time, the experiments were done, the partition coefficients between alkali chloride and alkali feldspar were not yet known. It was assumed that K0.95Na0.05Cl is close to the equilibrium composition of a melt coexisting with the sanidine. Partitioning coefficients determined by Neusser et al. (2012) imply that the equilibrium chloride composition for a sanidine with 85 mol% Or component is more sodic. However, as discussed below, this does not affect the results of this paper.
Analytics
After each experiment, the cuboid was cut in two different orientations, as shown in Fig. 1 of Behrens (2021b). After grinding and polishing to a thickness of about 500 µm, hydrogen profiles were recorded from the rim to the centre using an IR microscope A590 coupled with an FTIR spectrometer Bruker IFS88. A globar light source, a KBr beamsplitter and an MCT (mercury cadmium telluride) detector were applied in these measurements. 50—100 scans were accumulated for each spectrum with a spectral resolution of 2 cm−1. The spectrometer provides a partial polarized beam which is very well suited for precise measurements of diffusion profiles. A circular aperture corresponding to a focus diameter of 50 µm was applied, corresponding to an effective diameter of the exited area of 60 µm (Behrens 2021a). Some profiles were recently re-measured with an IR microscope Bruker IRscope II using a slit aperture to adjust an area of 20 × 100 µm2 in the focus plane. Aligning the long side parallel to the edge of the section, a spatial resolution of ≈ 40 µm could be achieved. The profiles measured by both setups show no significant difference.
Examples of IR absorption spectra of the sanidine in the range of OH stretching vibrations can be found in Behrens (2021a, b). The absorbance A of the main OH peak was determined after subtraction of a linear baseline. Hydrogen concentration was calculated by the Lambert–Beer law as described in these papers.
Results
Cuboids were well preserved after experiments with K-rich chlorides whilst pronounced crack formation in the near-surface region occurred in the runs with NaCl (SV-H1, SV-I1 and SV-FH). Cracks are aligned parallel to each other, but the orientation does not correspond to any of the feldspar cleavages. The lengths of the cracks depend on the orientation of the surface from which crack growth started, and for a given surface the cracks have all similar length (see Table 1). These observations agree well with those of Abart and co-workers (Neusser et al. 2012; Scheidl et al. 2013; Petrishcheva et al. 2019) who performed similar alkali exchange experiments with Eifel sanidines. In these papers, a detailed description of the mechanisms of alkali exchange and crack formation is given. The driving force for crack formation is the coherency stress created by the replacement of the large K+ ion by the smaller Na+ ions at the diffusion front.
Hydrogen profiles obtained from two experiments are presented in Fig. 1a, b. The complete set of experiments can be found in the electronic supplement. A common feature is that extrapolations of the measured profiles do not pass through the origin.
For one experiment using NaCl as the alkali source (SV-H1), a traverse from rim to the centre was analyzed by electron microprobe CAMECA Camebax. Measurement conditions were the same as reported in Behrens (2021a). The basic feldspar composition was preserved along the profile and only the Na/K ratio had changed. The alkali exchange profile was much shorter than the hydrogen profile (Fig. 2), and the crack tips evolved even beyond the region with measurable variation in alkali abundance.

Variation of alkali contents along the a axis after a desorption experiment in which a sanidine cuboid was embedded in NaCl powder. Lines illustrate the trend of the data. The sigmoidal shape of the curves is due to the concentration dependence of the Na–K interdiffusion coefficient (Christoffersen et al. 1983; Schäffer et al. 2014)
Discussion
Due to the limited spatial resolution of the IR beam, it is not possible to determine the concentration of hydrogen in the sanidine directly at the contact to the surrounding media. The first measurement point was typically 50 µm away from the surface and represents the average concentration over a range of 50 ± 30 µm. The measured profiles can be explained by a residual hydrogen content remaining in the surface area of the sanidine as well as by a very strong decrease of the concentration near the surface. Both approaches are discussed in following.
Profile evaluation assuming constant diffusivity
In the first approach, the absorbance–distance profiles were evaluated by the model of one-dimensional diffusion between an infinite reservoir (the gas phase) and a semi-infinite medium (the crystal) with the boundary condition of a constant surface concentration. Since the absorbance of the OH stretching vibration band is proportional to concentration of hydrogen, the measured absorbance can be directly used for the determination of the diffusion coefficient. Assuming a constant diffusion coefficient DH, the solution of Fick's second law for these boundary conditions is Crank (1975)
where t is the run duration, A is the absorbance at the distance x from the surface, Acenter and Asurface are absorbances in the centre and at the surface, respectively. The term DH characterizes the chemical diffusion of hydrogen during dehydrogenation. The measured profiles were fitted to Eq. 1 with Asurface, Acenter and DH as adjustable parameter. The fitted curves are shown as solid lines in Fig. 1a, b and fit results are also included in the electronic supplement. For all profiles, the correlation coefficient r2 of the fit was > 0.95, in many cases even > 0.99. This could be regarded as a good confirmation of the evaluation approach.
A unique feature of all experiments is that the fit curves do not pass through the origin, i.e. some hydrogen remained in the crystal at the contact to alkali chloride. The relative abundance of hydrogen at the surface, expressed by the absorbance ratio Asurface/Acenter, varies between 0.06 and 0.71. There is a rough trend of decreasing surface concentration of hydrogen with increasing temperature (Fig. 3). However, the scatter of the data is high, and the correlation coefficient is small (r2 = 0.45). One reason for the variation in Asurface/Acenter is that the water pressure (pH2O) in the experiments was low but it was not fixed at a constant value. Another reason is some variation in the hydrogen concentration in the centre of the sample between 81 and 115 ppm (see supplement). The trend of Asurface/Acenter with temperature is consistent with the structural interpretation of the type IIb OH defects in sanidine proposed in Behrens (2021a, b). Accordingly, this defect consists of a proton on an interstitial site compensating the charge deficiency of a nearby Al3+. Replacing hydrogen by a larger alkali requires energy for local expansion of the feldspar structure and, hence, substitution of hydrogen by alkali cations is a thermally activated process.

Variation of the hydrogen concentration at the surface of the crystals after desorption experiments with alkali chloride. The relative abundance of hydrogen at the surface compared to the centre, defined as the absorbance ratio Asurface/Acenter, was determined from fitting as shown in Fig. 1a, b. The dashed line is a guide for the eye to illustrate the evolution with temperature
Boltzmann–Matano (BM) analysis
In the second approach, it is assumed that the hydrogen content drops to zero at the surface during dehydrogenation. The strong decrease in hydrogen content towards the surface can be explained by a concentration-dependent diffusion coefficient. The method of Boltzmann (1894) and Matano (1932) allows determination of the diffusion coefficient for each concentration along a diffusion profile. For sorption and desorption profiles, the following equation can be used (Crank 1975):
where t is the time, C is the normalized concentration at the distance x from the surface, and C’ is the normalized concentration for which the diffusivity is evaluated. Considering that the IR absorbance is proportional to the concentration, in the case of desorption the normalized concentration is defined as
Thus, the normalized concentration varies between 1 at the surface (x = 0) and 0 at the end of the profile (x = ∞). It is more convenient to plot the concentration as a function of x (Behrens and Zhang 2009). Then Eq. (2) transforms to
Here x’ is the distance from the surface at which the normalized concentration equals C’. A graphical illustration of the evaluation method is given in Fig. 4. The dehydrogenation profiles in the sanidine can be fitted well with a polynomial Cnorm = 1 + a∙x + b∙x1.5 + c∙x2 + d∙x0.5 where a, b, c, and d are specific fit parameters for each profile. This type of equation has been successfully used to determine the concentration dependence of water diffusivity in silicate melts after dehydration experiments (Behrens 2006; Behrens and Zhang 2009).

Boltzmann–Matano analysis of dehydrogenation profiles. a Illustration of the method for desorption experiments. b Normalized concentration vs. distance for the example shown in Fig. 1a. c Normalized concentration vs. distance for the example shown in Fig. 1b. Lines in (b, c) represent fitting of the data with a polynomial Cnorm = 1 + a∙x + b∙x1.5 + c∙x2 + d∙x0.5 where a, b, c, and d are specific fit parameters. The basic assumption is that the surface concentration of hydrogen equals zero. The size of each symbol corresponds to its errors. Thin dashed lines mark the concentrations for which diffusion data are plotted in Fig. 6
Polynomial fitting is shown for two experiments in Fig. 4b, c. An upper value of x must be defined, since a good fit cannot be obtained with the polynomial for x → ∞. Artefacts easily occur in the BM method at low and at high concentrations when either slopes or integrals have high uncertainty. Therefore, only diffusion data for x ≥ 100 µm and Cnorm ≥ 0.1 are considered in following.
In Fig. 5, the diffusion coefficients obtained by BM analyses are plotted for the successful experiments. The general trend is an increase in diffusivity with increasing hydrogen concentration, expressed as A/Acenter. The curvature of the lines is not meaningful because it is strongly affected by imperfect polynomial fitting of the data. The increase in diffusion coefficients also supports the proposed defect model. The more protons have been replaced by alkalis, the greater the probability of a reverse reaction, i.e. migrating protons are bound locally again, reducing their overall mobility.

Diffusion coefficients as a function of the concentration of hydrogen expressed as the absorbance ratio A/Acenter, determined by the Boltzmann–Matano method. Only data for x ≥ 100 µm and A/Acenter < 0.9 are plotted
For both approaches, the temperature of diffusivity is well described by an Arrhenius law (Fig. 6a, b). Two selected values of c/ccenter, the diffusion coefficients are compared in Fig. 6b with the data of the first approach (constant diffusion coefficient) and the results of the D/H exchange experiments. The difference of diffusivities for c/ccenter = 0.9 and c/ccenter = 0.6 is about 0.25 log units, independent of temperature. The values for c/ccenter = 0.6 agree with the data obtained assuming constant proton diffusivity which demonstrates the consistency of both approaches. At first glance, it is surprising that diffusivities based on dehydrogenation for c/ccenter = 0.9 are higher by half a log unit compared to self-diffusivities of hydrogen determined by D/H exchange experiments with pre-annealed sanidines. Intuitively, one would expect the opposite, i.e. due to the temporary binding to dehydrogenated type IIb OH defects, the mobility of the protons is reduced.

Temperature dependence of chemical diffusion of hydrogen in sanidine at ambient pressure. a DH assuming constant diffusivity. b Chemical diffusivities of hydrogen for two different hydrogen contents (black solid lines) derived by the Boltzmann–Matano analysis. Dashed lines represent self-diffusivities of hydrogen determined by D/H exchange experiments in natural and heat-treated sanidine (Behrens 2021b). The pink line corresponds to the Arrhenius fit in (a)
However, the hydrogen concentrations measured by IR spectroscopy at room temperature only represent the stationary defects, but do not give any information about the concentration of mobile species (Kronenberg et al. 1996). As outlined by Behrens (2021b), the mobile hydrogen species in sanidines pre-annealed at ambient pressure are most likely protons while at elevated water pressures H2O molecules can enter the feldspar structure and became the main transporter for hydrogen. The concentration of mobile protons (cH+) is probably much lower than that of the hydrogen species bound in the stationary type II OH defects (cOH,II). The diffusion coefficient DD/H based on the hydrogen isotope exchange of the stationary OH defects is determined by the concentration ratio of the mobile and stationary hydrogen defects and the diffusivity of the mobile species DH+:
Assuming that DH+ is not significantly different for A/Acenter = 0.9 and A/Acenter = 1 (as adjusted in the pre-annealed sanidine), the difference between DH and DD/H simply reflects that finally 100% of the stationary OH defects are involved in isotope exchange, but only 10% in the dehydrogenation reaction at A/Acenter = 0.9. With increasing dehydrogenation, cH+ decreases due to the back reaction
where \({\text{Na}}_{{\text{OH,II}}}^{ + }\) is a sodium ion replacing a localized proton \({\text{H}}_{{\text{OH,II}}}^{ + }\) needed for local charge balance of excess aluminium. \({\text{Na}}_{i}^{ + }\) represents all kinds of sodium interstitials produced by the Frenkel equilibrium reaction
Here, the subscripts A refer to regular alkali sites in the feldspar structure and V indicates a vacancy.
The higher DD/H in the natural sanidine compared to the pre-annealed sanidine is due to the presence of water molecules in the former one. These molecules act as transport vehicles for hydrogen isotopes which can easily exchange with hydrogen in stationary defects. The higher diffusion coefficients for oxygen under hydrothermal conditions compared to dry conditions (see Fig. 7) also argue for such a mechanism at elevated water pressures.

Comparison of diffusion of hydrogen and oxygen species in feldspars. D/H interdiffusion is marked in black, H2O diffusion in blue and oxygen diffusion in red und proton diffusion in pink. Note that oxygen diffusion data are considered for Eifel sanidine only. For additional oxygen diffusion data and a detailed discussion, see the review of Farver (2010). Tracer diffusion data for Na and K in sanidine (in green) are shown to constrain the mechanism of proton removal. Data sources: [1] D–H interdiffusion, sanidine SV, pre-annealed, 1 atm, Behrens (2021b), [2] D–H interdiffusion,, sanidine SV, virgin, 1 atm, Behrens (2021b), [3] D–H interdiffusion, sanidine SV, pH2O = 2 kbar, Behrens (2021b), [4] D–H, adularia A1, pre-annealed, 1 atm, Behrens (2021b), [5] H2O desorption, plag An31, 1 atm, Johnson and Rossman (2013), [6] H2O desorption, adularia, 1 atm, Kronenberg et al. (1996), [7] H2O desorption, sanidine SV, virgin, 1 atm, Behrens (2021a), [8] H2O desorption, plag An66, 1 atm, Behrens (2021a), [9] H2O sorption, sanidine SV, pH2O = 6–8 kbar, this study, [10] 17O,18O diffusion, sanidine, pH2O = 1 kbar, Freer et al. (1997), [11] 18O diffusion, sanidine, 1 atm, dry, Derdau et al. (1998), [12] 22Na tracer diffusion, sanidine, 1 atm, dry, Wilangowski et al. (2015), [13] 43K tracer diffusion, sanidine, 1 atm, dry, Hergemöller et al. (2017), [14] Dehydrogenation supported by alkali, sanidine SV, pre-annealed, 1 atm, this study
Temperature dependence of hydrogen diffusion
Arrhenius parameters for dehydrogenation of sanidine are given in Table 2. Within error, the activation energies are identical for both evaluation approaches and do not depend on the degree of dehydrogenation, at least at high hydrogen contents. The activation energies for dehydrogenation of sanidine agree well with those for D/H exchange in natural Eifel sanidine (160.2 kJ/mol), pre-annealed Eifel sanidine (159.9 kJ/mole) and adularia from unknown locality (162.3 kJ/mole) reported in Behrens (2021b). A similar value of 172 kJ/mole is given by Kronenberg et al. (1996) for the removal of hydrogen defects in adularia from Kristallina, Switzerland. Kronenberg et al. (1996) suggest that proton migration via interstitials is the transport mechanism for hydrogen in their experiments. However, this interpretation is controversial, see the discussion by Doremus (1998), Kronenberg et al. (1998) and Behrens (2021b).
Much lower diffusivities at moderate temperatures and higher activation energies were found for dehydrogenation of andesine from Halloran Springs, California (Ea = 266–278 kJ/mole, Johnson and Rossman 2013) and Eifel sanidine in the absence of alkali chloride (Ea ≈ 305 kJ/mole, Behrens 2021a), see Fig. 7. These findings point to a different mechanism for removal of hydrogen defects. If the substitution of protons by monovalent cations or the release of hydrogen after oxidation of neighbouring ions such as Fe2+ is not possible, then oxygen must be additionally removed for charge compensation. This is associated with a strong perturbation of the crystal structure and thus requires high activation energies. In the case of dehydration of sanidine in absence of an external alkali source, such a mechanism is supported by cracking during complete hydrogen removal induced by internal strain (Behrens 2021a).
On the other hand, the activation energy appears to be significantly lower for D/H exchange under hydrothermal conditions (130 kJ/mole at a water pressure of 2 kbar, Behrens (2021b)). However, from the higher diffusivities and the low activation energies under hydrothermal conditions, one cannot necessarily conclude that water molecules have a higher mobility than protons in the feldspar structure. As already mentioned, the exchange rate is determined not only by the mobility of the defects but also by their concentration. In isotope exchange experiments at 6–8 kbar, an increase in hydrogen content of more than 50% was observed in some cases by Behrens (2021b). The incorporated water has a very high mobility, as shown by the chemical diffusion coefficients for H2O determined from the sorption profiles (see Fig. 7, data 9).
In Fig. 7, the diffusion data for hydrogen species are compared to alkali diffusion data for Eifel sanidine. It is striking that 22Na tracer diffusion (Wilangowski et al. (2015) are very similar to DH values determined by dehydrogenation of Eifel sanidine in presence of alkali chloride, while 43K tracer diffusion coefficients (Hergemöller et al. (2017) are several orders of magnitude smaller. My experiments with different alkali chlorides give consistent results which shows that the external source of alkali is not rate-controlling for dehydrogenation. The short Na/K exchange profiles when using sodium chloride as the alkali source support this thesis. As discussed in my previous paper (Behrens 2021a), the strongly bound type OH II defects in the sanidine are likely protons incorporated on interstitial sites providing charge balance for excess aluminium. Replacement by alkalis requires energy, and the smaller sodium ions are preferred over the larger potassium ions. Accordingly, the diffusion out of hydrogen requires a corresponding counterflux of sodium. However, since in the annealed sanidine the Na concentration is about 30 times higher than the H concentration, the local charge balance could be rapidly established even with an order of magnitude slower Na diffusivity.
Conclusions
Hydrogen defects can be incorporated as thermodynamically stable species in feldspars that crystallize at high temperatures in hydrous magmas. The nature of the defects and, hence, their stability in subsequent processes depend on the composition of the magmas. Thus, depending on the conditions, protons may be incorporated together with aluminium as a substitute for silicon, or water molecules may be associated with vacancies in the structure.
The new experiments presented here support the hypothesis that strongly bound, isolated OH defects in feldspars can be removed by diffusion of protons if alkalis are present for charge balance. If this is not the case, massive disruption of the crystal framework is required for dehydration, since oxygen ions must be removed in addition to protons to balance the charge. Under these conditions, the transport of hydrogen will preferentially occur by diffusion of neutral water molecules. If the defect centres are not irreversibly destroyed after hydrogen release, then they act as traps for protons and water molecules, respectively. As a result, the dehydration rate decreases with increasing degree of hydrogen removal.
The situation is different for feldspars in which hydrogen was incorporated under metamorphic conditions, for example during recrystallization. Here, the destruction of hydrogen defects is typically irreversible, and the mobility of released water molecules is not reduced by temporary binding in the defect centres. This appears to be the case with the Kristallina andesine studied by Kronenberg et al. (1996).
Data availability
All data are available via the electronic supplement.
References
Baschek G, Johannes W (1995) The estimation of NaSi-CaAl interdiffusion rates in peristerites by homogenization experiments. Eur J Mineral 7:295–307
Behrens H (2006) Water diffusion in silicate glasses and melts. Adv Sci Tech 46:79–88
Behrens H (2021a) Hydrogen defects in feldspars: defect properties and implications for water solubility in feldspar. Phys Chem Minerals 48:8
Behrens H (2021b) Hydrogen defects in feldspars: D/H interdiffusion and H2O diffusion in alkali feldspars. Phys Chem Minerals 48:27
Behrens H, Zhang Y (2009) H2O diffusion in peralkaline to peraluminous rhyolitic melts. Contrib Mineral Petrol 157:765–780
Behrens H, Zhang Y, Xu Z (2004) H2O diffusion in dacitic and andesitic melts. Geochim Cosmochim Acta 68:5139–5150
Beran A, LiIbowitzky E (2006) Water in natural mantle minerals II: olivine, garnet and accessory minerals. Rev Mineral Geochem 62:169–191
Boltzmann L (1894) Intergration der Diffusionsgleichung bei variablen Diffusions-koefficienten. Ann Phys 53:959–964
Cherniak DJ (2010) Cation diffusion in feldspars. Rev Mineral Geochem 72:691–733
Christoffersen R, Yund RA, Tullis J (1983) Interdiffusion of K and Na in alkali feldspars: homogenization experiments. Am Mineral 68(11–12):1126–1133
Crank J (1975) The mathematics of diffusion, 2nd edn. Clarendon press, Oxford, p 414
Derdau D, Freer R, Wright K (1998) Oxygen diffusion in anhydrous sanidine feldspar. Contrib Mineral Petrol 133:199–204
Doremus RH (1998) Comment on “Stationary and mobile hydrogen defects in potassium feldspar.” Geochim Cosmochim Acta 62:377–378
Elphick SC, Graham CM, Dennis PF (1988) An ion microprobe study of anhydrous oxygen diffusion in anorthite: a comparison with hydrothermal data and some geological implications. Contrib Mineral Pet 100:490–495
Farver JR (2010) Oxygen and hydrogen diffusion in minerals. Rev Mineral Geochem 72:447–507
Farver JR, Yund RA (1990) The effect of hydrogen, oxygen and water fugacity on oxygen diffusion in alkali feldspar. Geochim Cosmochim Acta 54:2953–2964
Freer R, Wright K, Kroll H, Göttlicher J (1997) Oxygen diffusion in sanidine feldspar and a critical appraisal of oxygen isotope-mass-effect measurements in non-cubic materials. Phil Mag A75:485–503
Giletti BJ, Semet MP, Yund RA (1978) Studies in diffusion-III. Oxygen in feldspars: an ion microprobe determination. Geochim Cosmochim Acta 42:45–57
Grove TL, Baker MB, Kinzler RJ (1984) Coupled CaAl–NaSi diffusion in plagioclase feldspar: experiments and applications to cooling rate speedometry. Geochim Cosmochim Acta 48:2113–2121
Hergemöller F, Wegner M, Deicher M, Wolf H, Brenner F, Hutter H, Abart R, Stolwijk NA (2017) Potassium self-diffusion in a K-rich single-crystal alkali feldspar. Phys Chem Miner 44:345–351
Johnson EA (2006) Water in nominally anhydrous crustal minerals: Speciation, concentration, and geologic significance. Rev Mineral Geochem 62:117–154
Johnson EA, Rossman GR (2003) The concentration and speciation of hydrogen in feldspars using FTIR and 1H MAS NMR spectroscopy. Am Mineral 88:901–911
Johnson EA, Rossman GR (2004) A survey of hydrous species and concentrations in igneous feldspars. Am Mineral 89:586–600
Johnson EA, Rossman GR (2013) The diffusion behavior of hydrogen in plagioclase feldspar at 800–1000 °C: implications for reequilibration of hydroxyl in volcanic phenocrysts. Am Mineral 98:1779–1787
Kronenberg AK, Yund RA, Rossman GR (1996) Stationary and mobile hydrogen defects in potassium feldspar. Geochim Cosmochim Acta 60:4075–4094
Kronenberg AK, Yund RA, Rossman GR (1998) Reply to the comment by Robert H. Doremus on “Stationary and mobile hydrogen defects in potassium feldspar.” Geochim Cosmochim Acta 62:379–382
Matano C (1932–3) The relation between the diffusion coefficients and concentrations of solid metals (the nickel-copper system). Jpn J Phys 8: 109–113
Neusser G, Abart R, Fischer F-D, Harlov D, Norberg N (2012) Experimental Na/K exchange between alkali feldspar and an NaCl-KCl salt melt: chemically induced fracturing and element partitioning. Contrib Mineral Pet 164:341–358
Petrishcheva E, Rieder M, Predan J, Fischer FD, Giester G, Abart R (2019) Diffusion-controlled crack propagation in alkali feldspar. Phys Chem Minerals 46:15–262
Schäffer A-K, Petrishcheva E, Habler G, Abart R, Rhede D, Giester G (2014) Sodium-potassium interdiffusion in potassium-rich alkali feldspar II: composition and temperature dependence obtained from cation exchange experiments. Am J Sci 314(9):1300–1318
Scheidl K, Schäffer A-K, Petrishcheva E, Habler G, Fischer F-D, Schreuer J, Abart R (2013) Chemically induced fracturing in alkali feldspar. Phys Chem Miner 41:1–16
Skogby H (2006) Water in natural mantle minerals I: pyroxenes. Rev Mineral Geochem 62:155–167
Wilangowski F, Abart R, Divinski SV, Stolwijk NA (2015) Radiotracer experiments and Monte Carlo simulations of sodium diffusion in alkali feldspar: evidence against the vacancy mechanism. Defect Diffus Forum 363:79–84
Yund RA (1986) Interdiffusion of NaSi-CaAl in peristerite. Phys Chem Mineral 13:11–16
Yund RA, Anderson TF (1974) The effect of fluid pressure on oxygen isotope exchange between feldspar and water. Geochim Cosmochim Acta 42:235–239
Acknowledgements
My special thanks go to Otto Dietrich for the excellent preparation of feldspar sections. I thank Fabian Hergemöller for stimulating thoughts on proton and alkali diffusion. Particular thanks to Andreas Kronenberg, Sylvie Demouchy and an anonymous third reviewer for fruitful comments. The research was supported by the researcher unit FOR2881 of the German Science foundation (DFG).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
I am the single author of the paper.
Corresponding author
Ethics declarations
Conflict of interest
The author declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Behrens, H. Hydrogen defects in feldspars: alkali-supported dehydrogenation of sanidine. Phys Chem Minerals 50, 18 (2023). https://doi.org/10.1007/s00269-023-01242-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00269-023-01242-9