
Abstract
HCN1 is one of four genes encoding hyperpolarization-activated cyclic nucleotide-gated channels. The phenotypic spectrum associated with HCN1 variants ranges from neonatal developmental and epileptic encephalopathy to idiopathic generalized epilepsy. We report a Japanese patient with repetitive focal seizures and super-refractory status epilepticus since early infancy caused by a de novo HCN1 variant, NM_021072.4, c.1195T>C, p.(Ser399Pro). This variant might have a dominant-negative effect on channel function, leading to severe epileptic encephalopathy.
Similar content being viewed by others
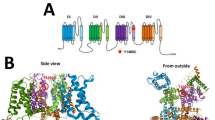

Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels mediate a cationic current that stabilizes the neuronal membrane potential against excitatory or inhibitory input and regulates neuronal network excitability1,2. HCN1 (NM_021072), one of the four genes that encode HCN channels1, is highly expressed in the neocortex, hippocampus, and brainstem of the central nervous system3. It was therefore presumed that pathogenic HCN1 variants could produce pharmacoresponsive epilepsy or developmental and epileptic encephalopathy (DEE). In 2014, Nava et al. identified five different de novo HCN1 variants in patients with Dravet-like syndrome without SCN1A and PCDH19 variants or fever-sensitive epileptic encephalopathy4. Additionally, recent studies have revealed that the phenotypic spectrum associated with HCN1 variants ranges from genetic epilepsy with febrile seizures plus (OMIM #618482) or genetic generalized epilepsy to neonatal- or infantile-onset DEE5,6,7,8,9,10 (OMIM #615871).
Herein, we report a Japanese patient with a de novo heterozygous HCN1 variant who had presented since early infancy with repetitive focal seizures and recurrent super-refractory status epilepticus, which were associated with profound developmental delay. This report provides detailed clinical findings that expand the known phenotypic spectrum of HCN1-related DEE.
The male patient, the second child of unrelated parents, was born at 39 weeks gestational age after a normal pregnancy and delivery. His birth weight, length, and occipitofrontal circumference were 3380 g (+0.8 standard deviation score [SDS]), 50.8 cm (+0.9 SDS), and 33.8 cm (+0.3 SDS), respectively. His father, elder brother, and maternal uncle had a history of febrile seizures. At 2 months of age, the patient developed convulsive seizures that gradually increased in frequency, occurring in clusters despite administration of phenobarbital and sodium valproate.
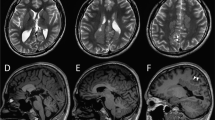
He was first admitted to our hospital at 4 months of age. His anthropometric measurements were as follows: body weight 7.6 kg (+0.6 SDS), height 60.5 cm (−1.6 SDS), and occipitofrontal circumference 42.4 cm (+1.3 SDS). Psychomotor development was normal. After admission, right or left hemi-clonic seizures and focal to bilateral tonic–clonic seizures were observed. Shortly afterward, a respiratory infection elicited a high fever and provoked the onset of convulsive status epilepticus. Intravenous anti-seizure medications, including diazepam, midazolam, phenytoin, and lidocaine, were ineffective. Ultimately, thiopental was effective in terminating seizure activity. An interictal electroencephalogram (EEG) showed spike discharges in the left frontal area. An ictal EEG revealed spike-wave discharge with onset in the left fronto-centro-parietal region (Fig. 1A) or right fronto-central region. Laboratory examinations and brain magnetic resonance imaging (MRI) findings showed no abnormalities at that time (Fig. 1B).

A An ictal EEG during right hemiclonic seizures at 4 months of age shows spike-wave discharges with onset in the left fronto-centro-parietal region. B Brain MRI at 4 months of age, showing normal findings. C At 8 months of age, after a second episode of convulsive status epilepticus, brain MRI shows severe frontal-dominant cortical and white matter atrophy with ventricular enlargement.
At 6 months of age, during another respiratory tract infection, the patient developed a prolonged generalized tonic convulsion lasting 30 min, which evolved into repetitive generalized convulsions with prolonged hypoxia. This prolonged seizure was refractory to anticonvulsants, including thiopental, and lasted 36 h. Pentobarbital was given intravenously, and mechanical ventilation was required for 2 weeks. After extubation, seizures were controlled by high-dose phenobarbital treatment combined with potassium bromide and zonisamide. However, the patient’s motor and cognitive development were severely impaired. Brain MRI at 8 months of age showed severe cortical and white matter atrophy (largely in the frontal lobe) and ventricular enlargement (Fig. 1C).
At 10 months of age, brief daily seizures involving grinning or a pale face were noted. At 13 years of age, the patient had generalized hypotonia and spastic quadriplegia with profound intellectual and motor impairment. He could not control his head, spoke no meaningful words and was almost entirely bedridden. An increase in brief seizures with tonic movements elicited the addition of levetiracetam and topiramate to existing medications (phenobarbital, potassium bromide, and zonisamide). However, these medications were only partially effective. At 18 years of age, the patient continued to have brief focal seizures, often in clusters requiring intravenous phenobarbital for seizure control.
Whole exome sequencing was performed using a SureSelectXT Human All Exon v5 (Agilent Technologies, Santa Clara, CA), and captured libraries were sequenced using an Illumina HiSeq 2500 (Illumina, San Diego, CA) with 101 base-paired end reads. Exome data processing, variant calling, and variant annotation were performed as previously described11. Variant pathogenicity was predicted using SIFT, Polyphen-2, CADD and M-CAP (Table S1). Whole exome sequencing of the patient’s leukocyte-derived genomic DNA identified an HCN1 variant: NM_021072.4, c.1195T>C, p.(Ser399Pro). Trio-based Sanger sequencing confirmed that this was a de novo variant. The variant was absent in 38 K JPN and gnomAD v2.1.1 (accessed on 22nd Feb 2023), as well as in our 408 in-house control exomes (all Japanese). The same variant was previously reported in a patient with infantile epileptic encephalopathy who had prolonged febrile seizures and intractable apneic tonic–clonic seizures starting at 4 months of age5. Therefore, the variant was classified as pathogenic according to the ACMG-AMP Guidelines (PS1, PS2, PM2, and PP3). The clinical and molecular genetic studies were performed in accordance with the Declaration of Helsinki and were approved by the institutional review board of Yamagata University Faculty of Medicine, Showa University School of Medicine, and Yokohama City University School of Medicine. The patient’s parents provided written informed consent.
We compared the features of the present patient with those of a previous patient carrying the same genetic variant5 and summarized the clinical manifestations of previously reported cases10 (Table 1). The present patient showed infantile-onset DEE with repetitive focal seizures, recurrent refractory status epilepticus, and progressive brain atrophy. Among previously reported patients with HCN1 variants, brain atrophy was observed in only one patient with a p.G391D variant5,10. Therefore, the phenotype in the present patient appears to be more severe than that of a previous patient with the same p.S399P variant. This phenotypic variability may be caused by genetic modifiers such as a prominent family history of febrile seizures even though no other variants in the known epilepsy-related genes were identified through whole exome sequencing of this patient. Progressive brain atrophy may also have been caused by acute encephalopathy or encephalitis. However, because MRI changes were not observed during the acute phase of status epilepticus in the patient, it was unlikely that he had acute encephalopathy or encephalitis.
Recurrent status epilepticus, refractory to intravenous anticonvulsant administration, was one of the characteristic features observed in this patient. Administration of general anesthesia was necessary to abolish a prolonged seizure lasting 36 hours. In previous studies, status epilepticus was reported in 5/18 patients with loss-of-function HCN1 variants and in 1/13 patients with gain-of-function HCN1 variants9. The p.S399P variant is also a loss-of-function variant. However, status epilepticus is caused not by a specific HCN1 variant but by a variety of variants9.
Both upregulation and downregulation of HCN channels have been associated with epileptic activity in animal models5,12. A previous study indicated that patients with the p.G391D variant showed the most severe phenotypes among HCN1 DEE5 patients, similar to the present patient with the p.S399P variant. HCN subunits have six transmembrane domains (S1–S6), including a positively charged voltage sensor (S4) and the ion-conducting pore region between S5 and S63. The amino acid glycine 391, located on the intracellular interface of the S6 transmembrane domain, is adjacent to the p.S399P variant5. Transient expression of HCN1 variants in Chinese hamster ovary cells showed that the protein levels of both the p.G391D and p.S399P variants were significantly decreased compared with those of wild-type proteins. In addition, whole-cell patch-clamp recordings showed no current in cells separately transfected with each variant, suggesting that these are loss-of-function variants5. Interestingly, when cells were cotransfected with wild-type and p.G391D variant constructs, a strong reduction in current density was observed, suggesting that p.G391D has a dominant-negative effect on heteromeric channel function. Similarly, severe DEE in the present patient may have been caused by a dominant-negative effect of the p.S399P variant.
The epileptic seizure frequency in patients with HCN1 variants ranges from no to daily seizures, and half of patients demonstrate resistance to anti-seizure medications5. High-dose phenobarbital and potassium bromide were partially effective for the present patient. In murine models of p.G391D and p.M153I, administration of the sodium channel antagonists lamotrigine and phenytoin resulted in paradoxical induction of seizures; however, administration of sodium valproate did not lead to convulsive seizures13. In Hcn1M294L mice carrying a homolog of the HCN1 p.M305L variant; phenytoin, lamotrigine, retigabine, and carbamazepine increased spike frequency, whereas levetiracetam, diazepam, sodium valproate, and ethosuximide significantly reduced spike frequency14. Sodium channel blockers such as carbamazepine or phenytoin may aggravate seizures in Dravet syndrome with SCN1A variants15. These blockers might also aggravate seizures in patients with HCN1 variants; therefore, early genetic diagnosis could be crucial for appropriate pharmacotherapy selection.
HGV database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3311.
References
Biel, M., Wahl-Schott, C., Michalakis, S. & Zong, X. Hyperpolarization-activated cation channels: from genes to function. Physiol. Rev. 89, 847–885 (2009).
Poolos, N. P. Hyperpolarization-activated cyclic nucleotide-gated (HCN) ion channelopathy in epilepsy. In: Noebels, J. L., Avoli, M., Rogawski, M. A., Olsen, R. W., & Delgado-Escueta, A. V. (eds). Jasper’s Basic Mechanisms of the Epilepsies, 4th edn. Oxford University Press: Oxford, USA, 85–96 (2012).
Benarroch, E. E. HCN channels: function and clinical implications. Neurology 80, 304–310 (2013).
Nava, C. et al. De novo mutations in HCN1 cause early infantile epileptic encephalopathy. Nat. Genet. 46, 640–645 (2014).
Marini, C. et al. HCN1 mutation spectrum: from neonatal epileptic encephalopathy to benign generalized epilepsy and beyond. Brain 141, 3160–3178 (2018).
Parrini, E. et al. Diagnostic targeted resequencing in 349 Patients with drug-resistant pediatric epilepsies identifies causative mutations in 30 different genes. Hum. Mutat. 38, 216–225 (2017).
Bonzanni, M. et al. A novel de novo HCN1 loss-of-function mutation in genetic generalized epilepsy causing increased neuronal excitability. Neurobiol. Dis. 118, 55–63 (2018).
Porro, A. et al. Do the functional properties of HCN1 mutants correlate with the clinical features in epileptic patients? Prog. Biophys. Mol. Biol. 166, 147–155 (2021).
Xie, C. et al. Novel HCN1 mutations associated with epilepsy and impacts on neuronal excitability. Front. Mol. Neurosci. 15, 870182 (2022).
Kessi, M. et al. The contribution of HCN channelopathies in different epileptic syndromes, mechanisms, modulators, and potential treatment targets: a systematic review. Front. Mol. Neurosci. 15, 807202 (2022).
Saitsu, H. et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 45, 445–449.e1 (2013).
Noam, Y., Bernard, C. & Baram, T. Z. Towards are integrated view of HCN channel role in epilepsy. Curr. Opin. Neurobiol. 21, 873–879 (2011).
Merseburg, A. et al. Seizures, behavioral deficits, and adverse drug responses in two new genetic mouse models of HCN1 epileptic encephalopathy. eLife 11, e70826 (2022).
Bleakley, L. E., McKenzie, C. E. & Reid, C. A. Efficacy of antiseizure medication in a mouse model of HCN1 developmental and epileptic encephalopathy. Epilepsia 64, e1–e8 (2023).
Wirrell, E. C. et al. International consensus on diagnosis and management of Dravet syndrome. Epilepsia 63, 1761–1777 (2022).
Acknowledgements
We thank the patient and his family for their participation in this study. This work was supported by the Japan Agency for Medical Research and Development (AMED) (grant numbers JP22ek0109486, JP22ek0109549, JP22ek0109493) and the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant-in-Aid for Scientific Research (B) (grant number JP20H03641) and (C) (grant number JH21K06819). We thank Edanz (https://jp.edanz.com/ac) for editing the English text of a draft of this manuscript.
Author information
Authors and Affiliations
Contributions
Y.K. cared for the patient and drafted the manuscript. N.A., K.Y., M.H., E.S., N.S., and T.O. cared for the patient. M.K. helped draft the manuscript and critically revised the manuscript for important content. M.N., H.S., and N.M. were responsible for genomic analysis, critically revised the manuscript for important content, and helped draft the manuscript. J.T. cared for the patient and revised the manuscript. All authors contributed to the writing of the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kobayashi, Y., Tohyama, J., Akasaka, N. et al. The HCN1 p.Ser399Pro variant causes epileptic encephalopathy with super-refractory status epilepticus. Hum Genome Var 10, 20 (2023). https://doi.org/10.1038/s41439-023-00247-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-023-00247-8
